





Abstract
CRISPR/Cas9 has dramatically changed how we conduct genetic research, providing a tool for precise sequence editing. However, new applications of CRISPR/Cas9 have emerged that do not involve nuclease activity. In the accompanying article “A dCas9-based system identifies a central role for Ctf19 in kinetochore-derived suppression of meiotic recombination,” Kuhl et al. utilize a catalytically dead Cas9 to localize proteins at specific genomic locations. The authors seek to understand the role of kinetochore proteins in the suppression of meiotic recombination, a phenomenon that has been observed in centromere regions. By harnessing the power of CRISPR/Cas9 to bind specific genomic sequences, Kuhl et al. localized individual kinetochore proteins to areas of high meiotic recombination and assessed their role in suppression. This primer article provides undergraduate students with background information on chromosomes, meiosis, recombination and CRISPR/Cas9 to support their reading of the Kuhl et al. study. This primer is intended to help students and instructors navigate the study’s experimental design, interpret the results, and appreciate the broader scope of meiotic recombination and CRISPR/Cas9. Questions are included to facilitate discussion of the study.
Introduction
The accuracy of chromosome segregation is particularly critical in meiosis, the specialized cell division that leads to the production of gametes. Incorrect chromosome segregation results in aneuploidy, the leading cause of infertility, miscarriage, and a significant source of birth defects. To promote proper segregation, meiotic cells create crossovers, sites of physical connection between homologous chromosomes. Crossovers hold chromosomes together until the proper time to separate and their resolutions facilitate exchanges of segments between chromosomes. These exchanges lead to new genetic combinations, critical for the evolution of new traits and species. In their article “A dCas9-based system identifies a central role for Ctf19 in kinetochore-derived suppression of meiotic recombination,” Kuhl et al. explored the regulation of crossovers and their associated recombination events using a novel application of the powerful genome editing tool CRISPR/Cas9. Kuhl et al. not only made important discoveries about crossovers but also established a new way to use CRISPR/Cas9, which can be applied to any study involving the precise location of proteins on chromosomes.
Background
Chromosome segregation
Chromosomes are the structures that contain our genetic information, and their proper separation in cell division is critical for the viability and reproductive success of an organism. Chromosomes consist of double-stranded DNA tightly wrapped around histone proteins. After DNA replication and chromosome condensation, identical sister chromatids are held together by protein rings called cohesins (Fig. 1a). Diploid organisms possess two copies of every chromosome, one inherited from each parent. These pairs are called homologous chromosomes, and they have the same size and gene location but can vary in the alleles they carry (Fig. 1b). Chromosomes are segregated during cell division, either mitosis or meiosis. In mitosis, cohesins are cleaved and sister chromatids are separated, resulting in two cells that each carry a complete set of chromosomes. During meiosis, however, diploid organisms reduce the copy number of each chromosome through two successive cell divisions, resulting in haploid cells that become gametes. In the first round of division (meiosis I), homologous chromosomes are separated (Fig. 1c), and in the second round (meiosis II), cohesins are cleaved and sister chromatids separate. If mistakes occur during the meiotic divisions, gametes can end up with missing or extra chromosomes, a condition called aneuploidy, which is a leading cause of infertility, miscarriage, and disorders such as Down syndrome, Patau syndrome, and Edwards syndrome (Nagaoka et al. 2012).
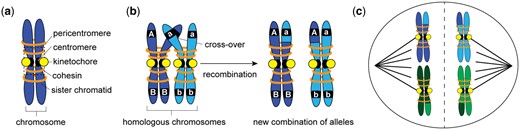
a) After replication, a chromosome consists of two sister chromatids held together by cohesin proteins. The centromere is bound by a protein structure called a kinetochore. In this study, Kuhl et al. investigated the ability of kinetochore proteins to suppress recombination; typically recombination is suppressed in the pericentromere, the chromatin region surround the centromere. b) In meiosis, homologous chromosomes pair and create crossovers. When crossovers between homologous chromosomes are resolved, segments of chromosomes are exchanged, and new genetic combinations are produced. The original haplotypes of the homologous chromosomes are AB and ab, but after recombination occurs at the cross-over location, two new combinations of alleles are created, Ab and aB. c) Crossovers hold pairs of homologous chromosomes together until they are separated in meiosis I by the action of the spindle, a microtubule-based machinery. The diagram here depicts the chromosome separation in meiosis I. In meiosis II, cohesin is degraded and sister chromatids are separated, resulting in haploid cells.
To segregate chromosomes during division, cells assemble a microtubule-based apparatus called the spindle that attaches to chromosomes through a protein structure called the kinetochore (Fig. 1c). Kinetochores vary in morphology across species, from a flat, disk-like structure in mammals to a “ball in a cup” structure in plants. Despite this variation, all kinetochores contain 50–100 different proteins organized into complexes that assemble on specific chromosome regions called the centromeres. Most multicellular organisms have large, regional centromeres that can span millions of bases, but simpler organisms, such as the budding yeast used by Kuhl et al., have centromeres that are only ∼125-bp long. One advantage of the budding yeast model for chromosome biology is the simplicity of the centromere and the known identity of all kinetochore proteins. Kinetochore proteins bind centromere sequences and attach to microtubules in the spindle, orienting chromosomes and facilitating their proper separation into two cells (Hinshaw and Harrison 2018).
Homologous chromosomes do not interact with one another except in early meiosis I. In mitotic cells, homologous chromosomes independently attach and align on the spindle, ultimately separating the sister chromatids. In meiotic cells, however, the homologous chromosomes must pair up to be correctly divided at the end of meiosis I. Synapsis, the zipping up of paired homologous chromosomes during meiosis I, is facilitated by a protein complex called the synaptonemal complex. The complex forms along the length of homologous chromosomes and holds them in positional alignment such that regions of sequence similarity are kept together (Gao and Colaiácovo 2018). In some species, synapsis of the homologs is required to proceed to meiotic recombination; however, in other species such as budding yeast, the initiation of meiotic recombination helps promote interaction between the homologs and assists the process of synapsis (Grey and de Massy 2022).
Crossovers and meiotic recombination
Meiotic recombination is a carefully regulated process of breaking DNA to promote the exchange of genetic material between homologous chromosomes. Crossovers are the sites of interaction between chromosomes, initiated with programmed breakage of both DNA strands (double-strand breaks, DSBs) (Lam and Keeney 2014). Their repair occurs through a process called homologous recombination that uses a DNA template. The template can either be a sister chromatid or the homologous chromosome. If the sister chromatid is used as a template to repair a DSB, no new combination of alleles is created as both chromatids are identical in DNA sequence. However, if the homologous chromosome is used as the repair template, a crossover is established and segments from the two homologous chromosomes can be exchanged, which can result in the creation of new combinations of alleles (or haplotypes) (Fig. 1b). The physical linkage of homologous chromosomes created by crossovers allows chromosomes to remain paired during spindle assembly and attachment. Crossovers are resolved at the moment of chromosome separation in meiosis I, and each homolog acquires new segments from the recombination event (Hunter 2015).
Meiotic recombination is essential for creating genetic diversity; it creates new combinations of alleles that serves as the genetic material on which evolution acts. For example, in Fig. 1b, the original haplotypes are AB on one homologous chromosome (dark blue) and ab on the other (light blue). Recombination between the two homologs created new haplotypes aB and Ab. These new combinations of alleles can provide advantages to individuals and over time can ultimately lead to new species. In this way, recombination is foundational to evolution broadly, and also specifically to animal and crop domestication, allowing humans to select useful combinations of traits. However, recombination does not occur evenly across the genome; some chromosomal locations, called hotspots, experience high levels of recombination, and others experience very little recombination (Peñalba and Wolf 2020). One region where recombination is suppressed is the centromere and the surrounding chromatin, called the pericentromere. Crossovers in the pericentromeric region could weaken cohesins, which hold sister chromatids together, or interfere with the kinetochore’s attachment to the spindle, both resulting in improper chromosome segregation and aneuploidy. As a result, organisms have evolved regulatory mechanisms to suppress recombination in the pericentromeric region (Kuhl and Vader 2019). Kuhl et al. (2020) investigated these mechanisms, specifically the ability of certain kinetochore proteins to inhibit the formation of crossovers and suppress recombination.
Research question and rationale
In a previous study, this group explored the ability of kinetochore proteins to inhibit recombination in the pericentromeric region (Vincenten et al. 2015). They found that the Ctf19 protein complex is able to both inhibit DSBs and increase cohesins in the pericentromeric regions by recruiting the cohesion-loading complex, Scc2/Scc4. Cohesins are known to block crossovers from forming, thus inhibiting recombination (Nambiar and Smith 2018). In their new study “A dCas9-based system identifies a central role for Ctf19 in kinetochore-derived suppression of meiotic recombination,” Kuhl et al. (2020) investigated which specific protein in the Ctf19 complex is responsible for the suppression of recombination. To identify individual protein function, the group placed different proteins from the Ctf19 complex at a location far from the kinetochore and pericentromeric region and measured their ability to suppress recombination at that site (Fig. 2a). This approach required the ability to control the placement of a protein at a specific genomic location without disrupting the DNA sequence. To accomplish this specific protein localization, the authors developed a new application for the CRISPR/Cas9 gene editing technology (Fig. 2b). Their study not only elucidates mechanisms for meiotic recombination suppression but also establishes a new way to use CRISPR/Cas9 to study chromosome biology.
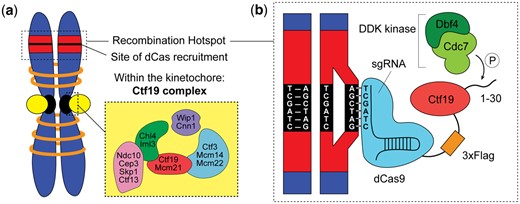
a) Within the larger kinetochore are many protein complexes, including the Ctf19/CCAN complex investigated by Kuhl et al., displayed in the zoomed in box. Within this complex are 5 distinct sub-complexes, each consisting of 2–5 individual proteins. Kuhl et al. initially screened one protein from each sub-complex (Ndc10, Iml3, Wip1, Ctf3, and Ctf19) for the ability to suppress recombination by targeting it to a recombination hotspot (red region) using CRISPR-dCas9. b) To target the individual kinetochore protein, Kuhl et al. designed a sgRNA that bound a specific genomic location (black region) within a recombination hotspot (red region). The sgRNA binds dCas9, which is fused to the kinetochore protein of interest. The diagram depicts the fusion of the Ctf19 to dCas9, along with a 3xFlag domain to facilitate biochemical experiments. Kuhl et al. demonstrated that Ctf19’s ability to suppress recombination is based on the phosphorylation of its N-terminal tail (1–30) by the DDK kinase. Note: this diagram shows a simplified view of the sgRNA with 6 nucleotides binding genomic sequence. In reality, sgRNAs are designed with 20 nucleotides of homology to a genomic sequence.
Tools and techniques
Yeast as a model system
Meiotic recombination occurs in all sexually reproducing species and the process is thought to be highly similar due to the conservation of proteins responsible for inducing DSBs. Homologs of these proteins are found across kingdoms, from yeast to mammals, and many studies in one species have informed discoveries in others (de Massy 2013). Kuhl et al. performed their study in Saccharomyces cerevisiae, also known as budding yeast or baker’s yeast. S. cerevisiae is a powerful model organism because it provides methods for creating transgenic strains as well as the simplicity of a single-celled microorganism; its population doubles every 90 min and it can be easily cultured and frozen for long-term storage (Duina et al. 2014). Nonetheless, budding yeast is a eukaryote, possessing membrane-bound organelles, linear chromosomes with centromeres, and microtubule-based spindle machinery.
Budding yeast are also capable of sexual reproduction. They typically exist as diploids, which reproduce asexually by budding identical cells, but diploids can be stimulated via nutrient starvation to undergo meiosis and produce 4 haploid cells. The 4 haploid cells, called spores, are contained within a structure called a tetrad. Because all 4 products of meiosis remain together, scientists are able to track all resulting genotypes from recombination through tetrad analysis. After meiosis is complete, 2 haploid cells can fuse to form a new, genetically distinct diploid cell. For further information about the budding yeast life cycle and research techniques, including tetrad analysis and making transgenics, see the primer review article by Duina et al. (2014). Kuhl et al. made use of the meiotic ability of budding yeast, as well as its genetic versatility and well-mapped genome to conduct their investigation.
Yeast nomenclature and convention are important to keep in mind while reading this article. Wild-type genes, genomic locations, and genetic constructs are designated with uppercase, italicized letters and numbers (e.g. CTF19, CEN8, 3xFLAG-dCAS9). Gene mutants are designated with lowercase, italicized letters and numbers (e.g. ctf19Δ, ctf191-30). Proteins and fusion proteins are designated with a starting uppercase letter, followed by lowercase letters, and are not italicized (e.g. Ctf19, Ctf19-3xFlag-dCas9).
CRISPR/Cas9 and protein targeting
Kuhl et al. developed a new application of CRISPR/Cas9 to investigate the role of individual kinetochore proteins in suppressing meiotic recombination. CRISPR/Cas9 is the revolutionary technology that has opened the door to precise DNA editing. With this tool, scientists can add, delete, insert, or modify any DNA sequence in any organism (Adli 2018). In recognition of the profound nature of its impact, the Nobel Prize was recently awarded to the scientists who pioneered its use, Jennifer Doudna and Emmanuelle Charpentier. At the heart of CRISPR/Cas9 is a specially designed RNA called a single-guide RNA (sgRNA) that brings the Cas9 nuclease to the desired DNA sequence. The sgRNA is comprised of two sections, the tracrRNA hairpin loop that binds Cas9 and the crRNA sequence designed to be complementary to the target DNA sequence. The sgRNA scans the genome, annealing to the target sequence and recruiting Cas9 to that location. In the original version of CRISPR/Cas9, the Cas9 nuclease cleaves the target sequence to induce editing (Thurtle-Schmidt and Lo 2018).
Kuhl et al. made use of the targeting capability of CRISPR/Cas9 but did not employ its editing function. To do this, they used a version of Cas9 that is deactivated or catalytically dead (dCas9) and unable to cleave DNA. The dCas9 protein is fused to each of the kinetochore proteins of interest and paired with a sgRNA designed to find a specific genomic location. The kinetochore protein is then targeted to an ectopic site, a location where it does not normally bind (Fig. 2b). In the Kuhl et al. study, the authors individually fuse Ctf19 complex proteins (Ctf19, Iml3, Wip1, Ndc10, and Ctf3) to dCas9 and confirm that these fusions are still functional proteins. The genomic locations chosen for the study are recombination hotspots, sites where recombination is known to occur at a high frequency. The authors designed 3 sgRNAs: (1) III, which binds 1.8 kb away from a hotspot on chromosome III, (2) VIII, which binds 2.5 kb away from a hotspot on chromosome VIII, and (3) a mock sgRNA, which does not have a crRNA sequence, thus does not bind DNA and, instead, serves as a negative control. By pairing these sgRNAs in combination with dCas9 fused to a kinetochore protein, the authors are able to target these proteins to the specific locations and determine their effect on meiotic recombination.
Measuring recombination frequency
The main assay used in Kuhl et al. is a measurement of recombination frequency at the hotspots targeted by the sgRNA-dCas9. When crossovers are formed, DNA sequences are swapped between homologous chromosomes and new allele combinations are created. In Fig. 1 of Kuhl et al., the authors describe their assay to measure recombination at the chromosome VIII hotspot. One copy of chromosome VIII contains the RFP gene, which produces a red fluorescent protein and the other chromosome VIII copy carries the GFP gene which produces a green fluorescent protein. If no recombination occurs during meiosis, no DNA is exchanged between chromosomes, and the resulting haploid cells will contain either RFP or GFP and thus be red or green in color. However, if recombination does occur, one haploid cell will be produced with a recombinant chromosome VIII that has both RFP and GFP and thus the cells will appear yellow. Another resulting haploid cell will have a recombinant chromosome VIII with neither fluorescent gene and will thus appear colorless. By culturing large numbers of yeast cells through meiosis and scoring the color phenotypes of many resulting haploid cells, Kuhl et al. could measure how frequently recombination occurs to produce these new recombinant genotypes. The occurrence of many yellow and colorless haploid cells in a population of yeast is indicative of a high recombination frequency.
Recombination frequency is reported in centimorgans, which is a measure of how frequently recombination occurs. It can also be used to reflect distance on a chromosome. Historically, scientists used recombination to help map the location of genes on chromosomes and determine their relative location. The further apart genes are on a chromosome, the higher the recombination frequency between them. The closer together genes are, the less likely a crossover is to form between them. The authors in this study assayed the recombination frequency on chromosome VIII between the RFP and GFP genes to determine if targeting proteins from each of the 5 Ctf19 sub-complexes (Fig. 2a, yellow box) impacts the rate of recombination at the hotspot. To confirm that the observed impacts are not specific to just chromosome VIII, Kuhl et al. also measured recombination at another genomic location on chromosome III using two different genes, the MAT gene that assists with sexual reproduction and the HIS3 gene that helps produce the amino acid histidine.
Using the data from their recombination assays, Kuhl et al. performed Fisher’s exact tests to determine whether recombination frequencies between groups of cells were significantly different. In this test, the null hypothesis is that the recombination frequencies between groups are not different. The P-value resulting from the Fisher’s test calculation is the probability of obtaining the experimentally derived results, assuming the null hypothesis is true. If the P-value is very low, it is unlikely that the difference in recombination frequency is due to this chance event; thus, the authors can reject the null hypothesis and conclude that the recombination frequencies in those groups are significantly different. Kuhl et al. used their recombination frequency assay not only to determine which kinetochore protein is responsible for meiotic suppression but also to further distinguish between different kinetochore protein.
Protein expression and localization: western blots, chromatin immunoprecipitation, and coimmunoprecipitation
In the study, it was critical to demonstrate that the dCas9-kinetochore fusion proteins were expressed and localized to the desired genomic locations. Kuhl et al. used western blots to validate the presence of the fusion proteins (Fig. 2b). In western blot analysis, cells are lysed and all cellular proteins are loaded into a gel to separate them by size and are then transferred to a membrane. The membrane is treated with antibodies that bind to a specific protein of interest, thus indicating that the protein is present. To assist with detection, proteins of interest are often tagged with small amino acid sequences called epitopes. These epitopes, such as FLAG and HA, allow scientists to use readily available antibodies for detection rather than create a custom antibody, which can be time consuming, expensive, and often fails. In Kuhl et al., the authors created a tripartite genetic construct that contained dCas9 fused to an epitope sequence (3xFLAG), fused to the kinetochore protein. In the western blot analysis, the authors used an antibody to FLAG (ɑ-FLAG) to detect the presence of their dCas9-kinetochore fusion protein.
To demonstrate that their dCas9-kinetochore proteins were localizing at the desired genomic location—a site separate from the centromere—Kuhl et al. used chromatin immunoprecipitation (ChIP). ChIP assays are used to identify the location of an interaction between a particular protein and chromatin. Chromatin is extracted from cells without removing bound proteins, sheared into small fragments and then the fragments (still attached to the proteins) are isolated by binding to an antibody that recognizes a specific protein. The isolated protein of interest brings along the chromatin segment to which it binds, and this chromatin can be sequenced or amplified via PCR to determine its identity. In this way, Kuhl et al. were able to isolate their FLAG-tagged dCas9-kinetochore fusion protein and establish whether it had been localized to the chromosome III or VIII recombination hotspots. They also performed the ChIP assay on other kinetochore proteins tagged with HA to determine if they were also recruited to these ectopic kinetochore sites.
Kuhl et al. also performed an assay called coimmunoprecipitation, which is used to demonstrate interactions between proteins. When the authors wanted to determine if 2 different proteins interacted with one another, they would isolate 1 protein using a specific antibody (for example use ɑ-FLAG to isolate the Ctf19-3xFlag-dCas9 protein complex) and probe with a second antibody to determine if the other protein was bound to it (in this example, use ɑ-HA to determine if Mcm21-3HA is bound).
Unpacking the study
Kuhl et al. investigated the role of 5 kinetochore proteins located in the Ctf19 complex (Ctf19, Iml3, Wip1, Ctf3, Ndc10) in the suppression of meiotic recombination (Fig. 2a). The authors developed a novel application of the CRISPR/Cas9 method, using the precision of sgRNAs to bring individual kinetochore proteins fused to dCas9 to a specific genomic location (Fig. 2b). Previous protein localization approaches required introducing DNA-binding sequences, such as the lac operon, into the desired genomic location and expressing proteins fused with binding proteins, such as the lac repressor. These approaches pioneered the idea of controlled localization of protein on chromatin, including studies in yeast (Kiermaier et al. 2009; Lacefield et al. 2009), Drosophila (Mendiburo et al. 2011), chicken (Hori et al. 2013), and human cells (Gascoigne et al. 2011; Logsdon et al. 2015) that localized kinetochore proteins on chromosome arms to create “synthetic kinetochores.” These studies, however, required the introduction of foreign DNA, altering the local genomic sequence and potentially disrupting important chromatin-based activities. These approaches also localized many molecules of the protein, where the Kuhl et al. approach not only leaves the native sequence undisturbed but also allows a single molecule to be localized. Kuhl et al. used this dCas9 tethering method to determine which kinetochore protein is responsible for meiotic suppression, as well as further dissect the specific protein domain and phosphorylation-based mechanism of suppression (Fig. 2b).
The novel CRISPR/Cas9 application developed by Kuhl et al. allowed them to modulate recombination frequencies at specific genomic locations. Further mechanistic insight into the kinetochore-based suppression of recombination, as well as use of this targeting tool, will allow scientists to manipulate recombination frequencies in pericentromeric regions, and more broadly across the genome. This work may also contribute to developing methods to target recombination to specific sites in plant and animal genomes. Plant and animal breeders use recombination to create new combination of traits that are superior to those in existing breeds. However, since crossover events are not uniformly distributed along chromosomes, breeders often find that crossovers are lacking in the chromosome regions containing genes encoding traits of interest. The ability to directly target recombination will unlock these regions where recombination was previously suppressed, revolutionizing crop and livestock breeding. It will also provide tools for research into human reproductive disorders, and expand our knowledge of fundamental biological processes.
Discussion questions: concepts in chromosome biology
1. Why and how are the frequency of recombination and distance along a chromosome related? Why might the relationship between the two be inexact, specifically what could skew this relationship such that recombination frequency would not correlate directly to physical distance on a chromosome?
2. What is a recombination hotspot? Why would organisms evolve mechanisms to suppress recombination at some locations and promote recombination at others?
3. How is repairing a double-strand break with a sister chromatid as a template distinct from crossover formation? How are the results different when repairs occur with a sister chromatid vs. a homologous chromosome?
4. Why might the presence of cohesin promote double-strand break repair using a sister chromatid as a template rather than the homologous chromosome?
Discussion questions: experimental design
5. Kuhl et al. showed a diagram of their dCas9 genetic construction in Fig. 1b; why was it important that they used the pHOP1 promoter which expresses only in meiosis? What could be the consequence of using a ubiquitous promoter (a promoter that is on all the time)?
6. The main assay of this article is a fluorescence-based recombination assay shown in Fig. 1e. What are the original parental genotypes and what are the new recombinant genotypes? What color phenotypes do these genotypes give rise to? Why was the blue CFP marker used in this study?
7. Why was the experiment shown in Fig. 1g a critical control for the dCas9 targeting system? How are the 3 sgRNAs different? How were the authors able to confirm that these sgRNAs are able to bring dCas9 to a specific location?
8. What is the Ctf19 truncation (ctf191-30) mutant? How did the authors use it in Fig. 4 to reach the conclusion that Ctf19 alone suppresses recombination?
9. What is the ctf19-19A and the ctf191-30-9A mutant? Why would the authors swap the serines and threonines on the N-terminal tail of Ctf19 for alanines? What important function is blocked in this mutant?
10. Why did the authors test the ability of 2 copies of the Ctf19 N-terminal tail (ctf191-30 (2x)) to suppress recombination? Why did they propose this as a better representation of Ctf19’s natural capacity? What does Fig. 5 show about this construct’s ability to suppress recombination?
11. Why did the authors choose known recombination hotspots to target the kinetochore proteins? How would this help the analysis of recombination suppression?
12. What experiment(s) could Kuhl et al. perform to strengthen their proposed Ctf19 mechanism? How could they demonstrate that the suppression of recombination at the ectopic location is caused by Ctf19-dCas9 directly recruiting Scc2–Scc4? How could they demonstrate that cohesin is enriched at the ectopic location?
Discussion questions: interpreting results
13. Kuhl et al. investigated 5 kinetochore proteins in the Ctf19 complex for their potential role in suppression of meiotic recombination.
What are the 5 kinetochore proteins?
Which protein(s) produced a suppressive effect on recombination?
Which figure reveals this result and what is being measured?
14. In this study, the authors fused kinetochore proteins to dCas9 to facilitate their localization to a recombination hotspot.
How can the authors be sure that dCas9 itself did not suppress recombination?
What control did they use to ensure the suppression was caused by the kinetochore protein and which figure shows this data?
15. The method used by Kuhl et al. is meant to bring a protein to a desired genomic location through the action of the sgRNA, and ultimately impact recombination at that location.
How did the authors demonstrate that protein localization was specific to the region specified by the sgRNA sequence?
What control experiments did the authors perform to demonstrate that recombination is impacted only at the site of protein localization?
Why were both a Ctf19-dCas9 construct and a dCas9 construct used in the experiment shown in Fig. 2d?
Why does the Ctf19-dCas9 construct suppress recombination when paired with the VIII gRNA but not when paired with the mock or III gRNA?
16. Previous work in budding yeast had shown that tethering a single kinetochore protein is sufficient to recruit multiple other proteins (Kiermaier et al. 2009; Lacefield et al. 2009); thus, Kuhl et al. wanted to determine if other kinetochore proteins were recruited to the ectopic site. What other proteins did the authors suspect were recruited to the ectopic Ctf19-dCas9 site and why?
17. Kuhl et al. found that their Ctf19-dCas9 protein fusion interacted with Mcm21 and Chl4.
How can this interaction be determined from the Western blots in Fig. 3, b and d? Which lane on the gel shows the interaction between the 2 proteins?
What do the ɑ-Flag and ɑ-HA antibodies probe for?
18. The western blot results from Fig. 3d show an interaction between Ctf19 and Chl4 that seems to conflict with the ChIP results from Fig. 3e.
Where is Chl4 located according to the ChIP data?
How is it possible that Ctf19-dCas9 and Chl4-3HA interact, but Chl4-3HA does not appear at the chromosome VIII location?
19. In Fig. 3c, what can we determine about the location of Mcm21-3HA? Why is Mcm21 located at chromosome VIII in only 1 experimental condition? What is this condition?
20. From the data shown in Fig. 4d, is the Ctf19-Mcm21 interaction dependent on phosphorylation?
21. The authors investigated the N-terminal tail of Ctf19 because previous studies had suggested that this domain is critical for interactions with Scc2–Scc4, a complex that suppresses recombination through loading of cohesin (Hinshaw et al. 2017).
Is the entire Ctf19 protein necessary to suppress recombination?
Does phosphorylation of the N-terminal tail of Ctf19 impact its ability to suppress recombination?
22. In Fig. 5, Kuhl et al. fused Dbf4, a subunit of the DDK kinase responsible for phosphorylating Ctf19, to dCas9 along with Ctf19.
What effect did this additional protein fusion have on recombination?
What important control did Kuhl et al. include in the recombination assay display in Fig. 5H?
Why did the authors suspect fusing Dbf4 to the Ctf19-dCas9 would alter recombination?
23. Why was the recombination assay in Fig. 6 needed? What additional information did Fig. 6, a–d provide to build the authors’ model of meiotic suppression?
24. Kuhl et al. propose 2 different mechanisms to suppress recombination: inhibiting double-strand breaks or promoting repair of double-strand breaks using a sister chromatid template. Figure 6 supports one of these 2 mechanisms:
What data are presented in Fig. 6E? Why is it necessary to perform this experiment in a saeΔ yeast strain?
Figure 6F is a quantification of the raw data shown in Fig. 6E. What conclusions can be drawn from this figure? What controls were used to help the authors reach this conclusion?
25. As the final experiment, Kuhl et al. demonstrated that the phosphorylation-based Ctf19 mechanism is important for recombination suppression in the pericentromeric region.
In Fig. 7, why do the authors use the cft19-9A and ctf19Δ mutant yeast strains? Are they expecting an increase or decrease in recombination in the pericentromeric region with these strains? Why?
Why is there a difference in recombination in the pericentromeric region but no difference in recombination on the chromosome arm in the different strains?
Why was this experiment critical to prove their model, and how was it different from their previous recombination assays?
Technical glossary
Centromere: DNA region located on chromosomes where kinetochore proteins assemble. Centromeres in S. cerevisiae are called point centromeres; they are small, ∼125-bp segments of DNA with a conserved sequence. Other species have larger regional centromeres, which can be millions of bases long with repeating sequences within them.
Cohesin: protein that forms rings encircling sister chromatids and holds them together until mitosis or meiosis II. Cohesins are loaded onto chromatids by the Scc2–Scc4 protein complex and have been shown to inhibit crossovers between homologous chromosomes.
CRISPR/Cas9: genomic engineering technology that allows scientists to precisely edit DNA. It requires (1) an sgRNA complementarily designed to base pair with a desired genomic location and (2) Cas9, a nuclease that cleaves DNA to induce sequence editing. In this study, the authors used a noncleaving version of Cas9 (dCas9) and fused it to kinetochore proteins they wished to localize to specific genomic locations.
dCas9: a catalytically dead version of Cas9 not capable of cleaving DNA but still able to bind the sgRNA and localize to a precise genomic location.
Ectopic: abnormal place, position, or activity. In this study, the authors used CRISPR/Cas9 to ectopically place a kinetochore protein at a genomic location where it normally does not reside.
Homologous chromosomes: chromosomes that possess the same size and genes, but they originate from different parents and often contain different alleles. Homologous chromosomes physically interact during meiosis I, being held together via crossovers that form between them.
Kinetochore: a large protein complex consisting of 50–100 proteins organized into smaller complexes such as the Ctf19 complex. Kinetochores bind to centromeres and attach chromosomes to microtubules in the spindle to facilitate proper chromosome segregation.
Pericentromeric region: chromatin region surrounding the centromere. In S. cerevisiae, the pericentromeric region is the 10–15 kb surrounding the centromere and it has been shown to have specialized 3D structure, high levels of cohesin and dense chromatin packaging.
sgRNA: sgRNA that consists of 2 components, a crRNA sequence that is specifically designed to be complementary to, and thus bind, a desired genomic sequence, and a tracrRNA sequence that folds into a hairpin loop and binds Cas9.
Spindle: microtubule-based apparatus that directs the movement of chromosome separation during cell division.
Data availability
No new data were generated or analysed in support of this research.
Acknowledgments
The authors would like to thank the Biology Department, the Arthur Levitt Public Affairs Center, and the Dean of Faculty at Hamilton College for their support through the Social Innovation and Transformative Leadership development grant.
Funding
This work is supported by the National Science Foundation: grant IOS-1848788.
Conflicts of Interest
The authors declare that there is no conflict of interest.

{kind=link}
{kind=link}