





Abstract
Nab2 encodes the Drosophila melanogaster member of a conserved family of zinc finger polyadenosine RNA-binding proteins (RBPs) linked to multiple steps in post-transcriptional regulation. Mutation of the Nab2 human ortholog ZC3H14 gives rise to an autosomal recessive intellectual disability but understanding of Nab2/ZC3H14 function in metazoan nervous systems is limited, in part because no comprehensive identification of metazoan Nab2/ZC3H14-associated RNA transcripts has yet been conducted. Moreover, many Nab2/ZC3H14 functional protein partnerships remain unidentified. Here, we present evidence that Nab2 genetically interacts with Ataxin-2 (Atx2), which encodes a neuronal translational regulator, and that these factors coordinately regulate neuronal morphology, circadian behavior, and adult viability. We then present the first high-throughput identifications of Nab2- and Atx2-associated RNAs in Drosophila brain neurons using RNA immunoprecipitation-sequencing (RIP-Seq). Critically, the RNA interactomes of each RBP overlap, and Nab2 exhibits high specificity in its RNA associations in neurons in vivo, associating with a small fraction of all polyadenylated RNAs. The identities of shared associated transcripts (e.g., drk, me31B, stai) and of transcripts specific to Nab2 or Atx2 (e.g., Arpc2 and tea) promise insight into neuronal functions of, and genetic interactions between, each RBP. Consistent with prior biochemical studies, Nab2-associated neuronal RNAs are overrepresented for internal A-rich motifs, suggesting these sequences may partially mediate Nab2 target selection. These data support a model where Nab2 functionally opposes Atx2 in neurons, demonstrate Nab2 shares associated neuronal RNAs with Atx2, and reveal Drosophila Nab2 associates with a more specific subset of polyadenylated mRNAs than its polyadenosine affinity alone may suggest.
Introduction
Intellectual disability refers to a broad group of neurodevelopmental disorders affecting approximately 1% of the world population (Maulik et al. 2011) and defined by significant limitations in intellectual functioning and adaptive behavior (Tasse et al. 2016; Vissers et al. 2016). Intellectual disabilities are etiologically diverse, and in some cases genetically complex, yet many exhibit overlapping molecular dysfunctions in a relatively limited set of fundamental neurodevelopmental pathways (reviewed in Chelly et al. 2006; van Bokhoven 2011; Verma et al. 2019). Thus, monogenic intellectual disabilities represent experimentally tractable avenues for understanding both these disorders more broadly and neurodevelopment in general (Najmabadi et al. 2011; Agha et al. 2014). One set of such informative monogenic intellectual disabilities is caused by mutations affecting genes encoding RNA-binding proteins (RBPs) (reviewed in Bardoni et al. 2012) such as ZC3H14 (zinc finger CCCH-type containing 14). Specifically, loss-of-function mutations in ZC3H14, which encodes a ubiquitously expressed zinc finger polyadenosine RBP, cause a nonsyndromic form of autosomal recessive intellectual disability (Pak et al. 2011; Al-Nabhani et al. 2018). However, the molecular functions and developmental roles of human ZC3H14 are largely unknown; defining these functions and roles provides an opportunity to better understand intellectual disability and human neurodevelopment.
Drosophila melanogaster has proved a powerful model system to understand the molecular functions of proteins encoded by many intellectual disability genes (Inlow and Restifo 2004; Oortveld et al. 2013), and ZC3H14 is no exception; the functions of ZC3H14 have begun to be dissected in part through study of the Drosophila ortholog Nab2 (Pak et al. 2011; Kelly et al. 2014). Drosophila Nab2, like ZC3H14, is a polyadenosine RBP that induces neurological defects when its expression is altered; deletion or overexpression of Nab2 causes neuronal morphological defects in the eye, axon projection defects in the developing brain, and memory impairments (Pak et al. 2011; Kelly et al. 2016; Bienkowski et al. 2017; Corgiat et al. 2021). The function of Nab2 is particularly important in Drosophila neurons, as pan-neuronal expression of Nab2 or an isoform of human ZC3H14 is sufficient to rescue the severe limitation in adult viability and locomotor defects caused by zygotic Nab2 deficiency (Pak et al. 2011; Kelly et al. 2014). Crucially, Nab2 physically and functionally interacts with Fmr1, the Drosophila homolog of the Fragile X Syndrome RBP FMRP (Verkerk et al. 1991; Ashley et al. 1993; Wan et al. 2000), to support axonal morphology and olfactory memory (Bienkowski et al. 2017). Studies suggest functions of Drosophila Nab2 in poly(A) tail length control, translational regulation, and proper mRNA splicing (Pak et al. 2011; Kelly et al. 2014; Bienkowski et al. 2017; Jalloh et al. 2021), but mechanistic demonstrations of the molecular function of Nab2 on individual, endogenous transcripts have yet to emerge. Such demonstrations have been prevented in large part because very few Nab2-associated RNAs have been identified (Bienkowski et al. 2017; Jalloh et al. 2021), and a comprehensive accounting of Nab2-associated RNAs in Drosophila neurons has yet to be conducted.
While the precise molecular function of Drosophila Nab2 on associated transcripts is unknown, informed hypotheses may be drawn by synthesizing research on Nab2 and orthologs murine ZC3H14, human ZC3H14, and Saccharomyces cerevisiae Nab2, the most well-studied Nab2/ZC3H14 ortholog (reviewed in Fasken et al. 2019). In S. cerevisiae, Nab2 functions pervasively across many RNAs in transcript stability and transcription termination, and likely acts similarly broadly in poly(A) tail length control and poly(A) RNA export (Anderson et al. 1993; Batisse et al. 2009; Schmid et al. 2015; Fasken et al. 2019; Alpert et al. 2020). Mutation of S. cerevisiae Nab2 induces dramatic increases in bulk poly(A) tail length and disrupts bulk poly(A) export from the nucleus (Green et al. 2002; Hector et al. 2002; Kelly et al. 2010). Consistent with its pervasive effects on many transcripts, S. cerevisiae Nab2 exhibits a broad binding target profile and is essential for cellular viability (Anderson et al. 1993; Tuck and Tollervey 2013). In contrast, mutant analyses of metazoan Nab2/ZC3H14 imply increased RNA target specificity for these proteins. Unlike Nab2 in S. cerevisiae, ZC3H14 in mice and humans is not essential for viability; instead, loss of ZC3H14 or Nab2 decreases viability in mice and flies and causes neurological or neurodevelopmental defects in both organisms (Pak et al. 2011; Rha et al. 2017b; Al-Nabhani et al. 2018; Wheeler et al. 2019). Bulk poly(A) tail lengths increase upon loss of Nab2 in Drosophila or ZC3H14 in mice, but this increase is not observed across all mouse tissues or all individual Drosophila mRNAs tested, and it is less pronounced than the effects observed in S. cerevisiae (Kelly et al. 2010; Bienkowski et al. 2017; Rha et al. 2017b). Moreover, in Drosophila and mouse cells, respectively, a pervasive nuclear poly(A) export defect is not observed upon Nab2 loss or ZC3H14 knockdown (Farny et al. 2008; Pak et al. 2011; Kelly et al. 2014). Drosophila Nab2 is required for proper splicing of individual introns and exons, but only for a small set of transcripts (Jalloh et al. 2021). Taken together, these data are consistent with a focused role for Drosophila Nab2 in regulating poly(A) tail length, splicing, stability, and/or nuclear export critical for specific transcripts, cell types, and/or developmental contexts (Bienkowski et al. 2017; Rha et al. 2017b; Jalloh et al. 2021). Crucially, however, the theme of Drosophila Nab2 RNA target specificity implied by these data has not been tested and remains an important open question, especially as the polyadenosine affinity of Drosophila Nab2 (Pak et al. 2011) makes Nab2 theoretically capable of associating with all polyadenylated transcripts via their poly(A) tails. Thus, a comprehensive identification of Drosophila Nab2-associated RNAs is necessary to determine the potential scope of Nab2 function and provide sets of transcripts on which the molecular consequences of Nab2–RNA association may be systematically evaluated. In the present study, we define the first neuronal RNA interactome for Nab2.
Contextualizing Nab2–RNA associations requires further definition of the molecular pathways and proteins, particularly other RBPs, that Nab2 interacts with or regulates. Notably, the Nab2 modifier eye screen that initially linked Nab2 and Fmr1 (Bienkowski et al. 2017) also recovered an allele of Ataxin-2 (Atx2), which encodes a conserved RBP and regulatory partner of Fmr1 in Drosophila neurons (Jimenez-Lopez and Guzman 2014; Sudhakaran et al. 2014). The shared connection of Nab2 and Atx2 with Fmr1 raised the possibility of cooperation or competition between these two proteins. Underscoring the value of this approach, Atx2 is a protein of particular importance for human health and neuronal function. Expansion of a polyglutamine tract within ATXN2, the human Atx2 ortholog, gives rise to the autosomal dominant neurodegenerative disease spinocerebellar ataxia type 2 (SCA2) (Imbert et al. 1996; Pulst et al. 1996; Sanpei et al. 1996). Expansions of the same tract are also associated with parkinsonism and amyotrophic lateral sclerosis (Gwinn-Hardy et al. 2000; Elden et al. 2010; Park et al. 2015). Functionally, Atx2 encodes a conserved RBP that regulates protein translation, mRNA stability, and mRNP granule formation and plays roles in memory, cellular metabolism, and circadian rhythms (reviewed in Ostrowski et al. 2017; Lee et al. 2018). Among the most well-studied molecular roles of Atx2 are its contributions to regulation of mRNA translation in the cytoplasm. Specifically, Atx2 suppresses the translation of some target RNAs through RNP granule formation and interactions with the RNA interference (RNAi) machinery (McCann et al. 2011; Sudhakaran et al. 2014; Bakthavachalu et al. 2018) and supports the translation of other targets by promoting RNA circularization (Lim and Allada 2013; Zhang et al. 2013; Lee et al. 2017). Intriguingly Atx2, like Nab2, contributes to poly(A) tail length control in S. cerevisiae—the yeast Atx2 homolog Pbp1 promotes poly(A) tail length, likely by inhibiting the activity of poly(A) nuclease (PAN) (Mangus et al. 1998, 2004). The shared connections of Nab2 and Atx2 to Fmr1, neuronal translation, and poly(A) tail length control emphasize the potential for and need to test whether these RBPs functionally interact beyond the initial eye screen link.
Here, after expanding the genetic link previously identified between Nab2 and Atx2 in the eye modifier screen (Bienkowski et al. 2017), we have used genetic and molecular approaches to probe the functional connections between these two RBPs. We show that Nab2 and Atx2 functionally interact to control neuronal morphology of the mushroom bodies (MBs), a learning and memory center of the Drosophila brain (Heisenberg 2003; Kahsai and Zars 2011; Yagi et al. 2016; Takemura et al. 2017), as well as to control circadian cycles in timeless-expressing neurons. We then couple these genetic data with the first high-throughput identification of Nab2- and Atx2-associated RNAs in Drosophila; in fact, this accounting has been performed for Nab2 only in S. cerevisiae (Kim Guisbert et al. 2005; Batisse et al. 2009; Tuck and Tollervey 2013; Baejen et al. 2014), not in many metazoan. This molecular approach reveals that Nab2 and Atx2 associate with an overlapping set of RNA transcripts in fly brains and provides insight into the possible functions of each protein individually and in concert with one another. As functional validation of these interactions, genetic tests confirm that loss-of-function alleles corresponding to a select subset of Nab2-associated mRNAs dominantly modify the Nab2-overexpression phenotype in the eye modifier screen. Considering these data as a whole, we propose a model in which the genetic interactions between Nab2 and Atx2 are explained by their counterbalanced regulation of shared associated RNAs. In sum, these data represent a valuable resource for understanding the neuronal roles of Nab2 and Atx2 in Drosophila and, potentially, for understanding links between each RBP and human disease.
Materials and methods
Drosophila genetics and husbandry
Genetic crosses of D. melanogaster were raised on standard media and maintained at 25°C in humidified incubators (SRI20PF, Shel Lab) with 12-h light–dark cycles unless otherwise specified. Cultures were often supplemented with granular yeast (Red Star Yeast) to encourage egg laying. Parental stocks were maintained at either at room temperature (RT) or 18°C to control virgin eclosion timing. Stocks used include Nab2ex3 (a Nab2 null), Nab2pex41 (a P-element excision control serving as a Nab2 wild type), and UAS>Nab2-FLAG, all first described in Pak et al. (2011). Additional stocks used include GMR-Gal4 (chromosome 2), per01 (gift of M. Rosbash), Atx2X1 (an Atx2 null, gift of N. Bonini) (Satterfield et al. 2002), and UAS>Atx2-3xFLAG (gift of R. Allada) (Lim and Allada 2013). Stocks sourced from the Bloomington Drosophila Stock Center (BDSC) include: elav>Gal4 (elavc155, BL458) (Lin and Goodman 1994), OK107-Gal4 (BL854) (Connolly et al. 1996), tim-Gal4 (BL80941) (Blau and Young 1999), Df(3R)Exel6174 (BL7653) (Parks et al. 2004), UAS>Nab2 (Nab2EP3716, BL17159) (Rorth et al. 1998; Bellen et al. 2004), mtd4 (BL4872), drke0A (BL5691), Df(3L)Exel6137 (BL7616), me31Bk06607 (BL10635), Bsgk13638 (BL11096), HmgZKG01188 (BL13704), Arpc2KG04658 (BL13978), staiKO (BL58438), GαoKO (BL81098), sm1 (BL400), and Atx2DG08112. The Atx2DG08112 stock (Huet et al. 2002) was mapped as part of the Gene Disruption Project (GDP) (Bellen et al. 2004) and is no longer available from the BDSC; copies provided upon request.
Drosophila eye imaging
Drosophila eyes were imaged using a Leica MC170 HD digital camera mounted on a Nikon SMZ800N stereo microscope at 8× magnification. To prepare subjects for imaging, flies were flash frozen (−80°C, 1 min), fixed in place on a clear Slygard pad using minutien pins (26002-10, Fine Science Tools), and submerged in 70% ethanol to diffuse light and reduce glare. Subjects were illuminated with a fiber optic ring light (Dolan-Jenner) and LED illuminator (Nikon Instruments, Inc.) and image acquisition was performed using the Leica Application Suite (v4.12) for Windows under the following parameters: 140 ms exposure; automatic white balance; highest available resolution; and default values for gain, saturation, gamma, and hue. Each subject was imaged at multiple focal planes (often ≥10), and these were subsequently combined using the Auto-Align and Auto-Blend functions and sharpened using the Smart Sharpen function in Photoshop CS5.1 Extended (Adobe) to generate final, merged images in which the entire subject is in-focus. These “focus stacking” processing steps (Patterson) combine only in-focus regions of an image series into a single, merged image.
Immunofluorescence
For MB morphology imaging, Drosophila brains were dissected using methods similar to those in Williamson and Hiesinger (2010), Kelly et al. (2016), and Kelly et al. (2017). Briefly, using #5 Dumont fine forceps (Ted Pella, Inc.), for each dissection a Drosophila head was isolated in PBS (often supplemented with 0.1% Triton X-100), the proboscis was removed to provide a forceps grip point, and the remaining cuticle and trachea were peeled away from the brain within. On wet ice, dissected brains were fixed in 4% paraformaldehyde for 30 min and then permeabilized in 0.3% PBS-Triton (PBS-T) for 20 min. For both primary and secondary antibody incubations, brains were left rocking at 4°C for one to three nights in 0.1% PBS-T supplemented with blocking agent normal goat serum (Jackson ImmunoResearch) at a 1:20 dilution. Immunostained brains were mounted on SuperFrost Plus slides (12-550-15, Fisher Scientific) in Vectashield (H-1000, Vector Laboratories) using a cover slip bridge (Kelly et al. 2017). Brains were imaged on a Zeiss LSM 510 confocal microscope. Exclusively female flies were dissected for practicality, given that Nab2ex3 nulls were analyzed in this experiment, and Nab2ex3 adult viability skews toward females (Jalloh et al. 2021).
For Nab2–Atx2 localization experiments, whole animals were fixed in 4% paraformaldehyde, 0.008% PBS-T, shaking, for 3 h at RT and then washed in PBS and stored at 4°C overnight. Brains were dissected in 0.008% PBS-T using similar methods as described above, permeabilized by shaking in 0.5% PBS-T overnight at 4°C, and blocked by shaking in 0.5% PBS-T, 5% NGS for 2 h at RT. For both primary and secondary antibody/Hoechst incubations, brains were left shaking at 4°C for two to three nights in 0.5% PBS-T, 5% NGS. After washing with 0.5% PBS-T followed by PBS, brains were mounted in SlowFade Gold Antifade Mountant (S36936, Invitrogen), surrounded by an adhesive imaging spacer (GBL654002, Sigma-Aldrich) to prevent sample compression, and finally cover-slipped and sealed with clear nail polish. Brains were imaged on an A1R HD25 confocal microscope (Nikon) and a multiphoton FV1000 laser-scanning microscope (Olympus).
Primary antibodies and dilutions used are as follows: mouse α-Fasciclin 2 (1:50) (1D4, Developmental Studies Hybridoma Bank), rabbit α-GFP (1:400) (A11122, Invitrogen), and mouse α-FLAG (1:500) (F1804, Sigma-Aldrich). Secondary antibodies and dilutions used are as follows: goat α-mouse Cy3 (1:100) (Jackson ImmunoResearch), goat α-mouse Alexa 594 (1:400) (A11032, Invitrogen), and goat α-rabbit Alexa 488 (1:400) (A11008, Invitrogen). To fluoresce DNA and mark nuclei in localization experiments, brains were also incubated with a Hoechst 33342 stain (1:1000) (H21492, Invitrogen) during secondary antibody incubation.
Further brain image analysis and processing, including generating maximum intensity projections and focus stacks and adjusting brightness and contrast, was performed with Photoshop CS5.1 Extended (Adobe) and Fiji (Schindelin et al. 2012), a distribution of ImageJ (Schneider et al. 2012; Rueden et al. 2017).
Circadian analysis
Adult male flies of each genotype were selected on the day of eclosion and aged for 2–5 days on Genesee Nutrifly MF food at 25°C on a 12-h light/dark cycle. Circadian parameters were then determined as in Geissmann et al. (2019a). Briefly, flies were loaded into individual polycarbonate tubes containing food, placed into Drosophila Activity Monitors (DAM, Trikinetics), and housed at 25°C. All flies were initially entrained using a 12 h of light/12 h of dark schedule. Following the third day of entrainment, lights were switched off and flies were then housed for an additional 7 days of complete darkness. The number of beam breaks per minute was collected for each fly during the entire 10-day period.
Following the last 24-h period of darkness, the percentage of rhythmic flies, the average circadian period length, and average period power was calculated for each genotype using the chi-squared periodogram analysis within the Rethomics R package as described (Geissmann et al. 2019b). Pairwise Fisher’s exact tests with Bonferroni correction for multiple comparisons were used to analyze % rhythmicity between genotypes. Nonparametric Kruskal–Wallis tests and pairwise Wilcoxon Rank Sum Tests with Bonferroni correction were performed to analyze differences in circadian period length and power between genotypes. R scripts are available upon request.
Immunoprecipitation
This immunoprecipitation protocol was developed through optimization guided by the protocols presented in Yang et al. (2005), Banerjee et al. (2017), Bienkowski et al. (2017), and Morris and Corbett (2018). Nuclear isolation buffer (NIB; 10 mM Tris–HCl pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40) and immunoprecipitation buffer (IP buffer; 50 mM HEPES, 150 mM NaCl, 5 mM EDTA, 0.1% NP-40) were prepared ahead of the experiment and stored indefinitely at 4°C. Both buffers, and the glycine and PBS solutions below, were prepared primarily in 0.1% diethyl pyrocarbonate (DEPC)-treated and autoclaved ultrapure Milli-Q water to limit RNase contamination. Both NIB and IP buffer were supplemented with an EDTA-free cOmplete protease inhibitor cocktail tablet (1 tablet/28 ml; 11873580001, Roche) and RNasin Plus RNase inhibitor (0.2%; N2615, Promega) freshly before each experiment. Additionally, before each experiment Protein G-coupled magnetic dynabeads (10003D, ThermoFisher) were conjugated to glycerol-free (Domanski et al. 2012) monoclonal α-FLAG (F3165, Sigma-Aldrich) in aliquots of 1.5 mg beads/9 µg antibody by incubation for 45 min at RT. Throughout the experiment, beads were magnetized using DynaMag-Spin magnets (e.g., 12320D, ThermoFisher) as necessary. Exclusively female flies were used for consistency with MB experiments and for practicality, as both elav>Nab2-FLAG and elav>Atx2-3xFLAG prohibitively decreased relative male viability (data not shown), presumably due to deleterious effects of enhanced Gal4 and epitope-tagged protein expression in males driven by dosage compensation of the X-chromosome-linked elav>Gal4 construct.
Three-hundred female Drosophila heads of each of the following genotypes elav>Gal4 alone, elav>Nab2-FLAG, and elav>Atx2-3xFLAG, previously isolated in bulk (see Supplementary Materials and Methods), were fixed in 1% formaldehyde, 0.1% NP-40 in PBS for 30 min at 4°C. Fixation was quenched by adding glycine to a final concentration of 250 mM and rocking for 10 min at 4°C. Heads were washed in 0.1% NP-40 in PBS and then manually homogenized with a smooth Teflon pestle for 5 min in 250 µl of NIB in a size AA glass tissue grinder at 4°C (3431D70, Thomas Scientific). Homogenates were spun through 35 µm cell strainer caps into round-bottom tubes (352235, Falcon) to remove exoskeletal debris, transferred, and then centrifuged for 5 min at 500 × g at 4°C to separate an insoluble fraction. Twenty percent of the soluble supernatant volume was isolated and defined as Input; the remaining 80% was used for immunoprecipitation. Both Input and IP samples were diluted to final concentrations of 0.8× IP buffer to ensure comparable and efficient sample lysis. IP samples were transferred onto the α-FLAG-conjugated magnetic Dynabeads, and both sample types were incubated, rotating, for 10 min at RT. Next, IP sample supernatant was collected as the unbound fraction, and IP sample beads were washed three times in IP Buffer. Finally, IP sample beads were resuspended in IP Buffer, transferred to clean tubes, and stored along with Input samples overnight at 4°C to allow passive hydrolysis to partially reverse formaldehyde crosslinks. This protocol was applied for both protein coimmunoprecipitation and RNA immunoprecipitation.
For protein coimmunoprecipitation, harsh elution of protein from IP sample beads was accomplished the next day; IP samples were diluted in modified Laemmli Sample Buffer (Laemmli 1970), incubated at 98°C for 5 min, centrifuged at 16,100×g for 5 min at RT, and magnetized to collect beads. Sample supernatants were then collected as IP samples. In parallel, Input samples were concentrated using an acetone-based method; this step was required for subsequent immunoblot analysis. Input samples were diluted to generate 80% chilled acetone solutions, vortexed for 15 s, and incubated at −20°C for 60 min. Samples were centrifuged at 14,000 × g for 10 min at RT, resulting supernatants were discarded, and most remaining acetone was evaporated by air drying protein pellets in open tubes for 30 s at RT. To solubilize these dried protein pellets, samples were suspended in a solution equal parts modified Laemmli Sample Buffer and IP Buffer, vortexed, sonicated for 3×5 min in a 4°C Bioruptor ultrasonicator (UCD-200, Diagenode), vortexed, and heated at 98°C for 10 min. Finally, remaining insoluble material was collected by centrifugation at 16,100 × g for 5 min at RT. Associated supernatants were isolated as concentrated Input protein samples. For RNA immunoprecipitation, harsh elution of RNA from IP sample beads was accomplished the next day with TRIzol. Both IP and Input samples were subjected to the RNA extraction protocol detailed below.
RNA extraction
Following immunoprecipitation, RNA was isolated from IP and Input samples using a TRIzol-column hybrid approach adapted from Dr Mauricio Rodriguez-Lanetty (see Acknowledgments). To account for volume differences, samples were vigorously homogenized in TRIzol reagent (15596018, ThermoFisher) at a ratio of either 1:10 (IP sample: TRIzol) or 1:3 (Input sample: TRIzol) and then incubated for 5 min at RT. All homogenized samples were clarified by centrifugation at 12,000×g at 4°C for 5 min, IP samples were magnetized to collect beads, and supernatant was isolated from all samples. After adding chloroform at a ratio of 0.2:1 (choloroform:TRIzol), samples were manually shaken and incubated at RT for 3 min. Samples were phase separated by centrifugation at 12,000×g at 4°C for 15 min, after which the aqueous layer was carefully isolated and mixed with an equal volume of 100% ethanol. RNA was further purified using an RNeasy Mini Kit (74106, QIAGEN) according to the manufacturer’s instructions (RNeasy Mini Handbook, 4th Ed., June 2012) with the following deviations: for each sample, a final 30 µl elution was performed twice, isolating 60 µl of RNA in total into each collection tube. An on-column DNase digestion step was also performed under the same instructions using an RNase-Free DNase Set (79254, QIAGEN). Final RNA concentration and sample purity were determined via a NanoDrop 1000 spectrophotometer (ThermoFisher).
RNA sequencing
RNA from twelve samples of 300 adult female Drosophila heads each was isolated via the immunoprecipitation and extraction protocols described above, generating 12 pairs of IP and Input samples, or 24 samples in total. These samples were composed of four biological replicates each of elav>Gal4 alone, elav>Nab2-FLAG, and elav>Atx2-3xFLAG. Once obtained, RNA samples were transferred on dry ice to the Georgia Genomics and Bioinformatics Core at UGA for library preparation and sequencing. There, IP samples were first concentrated using solid phase reversible immobilization beads. Then, the TruSeq Stranded Total RNA Library Prep Gold kit (20020598, Illumina) was used to deplete rRNA and prepare stranded cDNA libraries from all 24 samples. These uniquely barcoded cDNA libraries were then pooled by sample type, forming one IP library pool and one Input library pool. Each pool was sequenced on a separate NextSeq High Output Flow Cell (Illumina) for 150 cycles to generate paired-end, 75 base-pair (bp) reads. Total nonindex sequencing yield across all IP samples was 88.49 Gbp, equivalent to about 1.2 billion reads in total and 98 million reads per sample. Total nonindex sequencing yield across all Input samples was 83.25 Gbp, equivalent to about 1.1 billion reads in total and 93 million reads per sample. Sequencing accuracy was high; 87.83% and 91.38% of nonindex reads for IP and Input samples, respectively, have a sequencing quality (Q) score greater than or equal to 30.
RNA sequencing analysis—read mapping, differential expression, and visualization
Following sequencing, raw read FASTA files were transferred to Emory for bioinformatic analysis. To start, analyses were conducted on the Galaxy web platform, specifically using the public server at usegalaxy.org (Afgan et al. 2018). This analysis was supported by the BDGP6.22 release of the D. melanogaster genome (Hoskins et al. 2015). Both the raw sequence FASTA and the gene annotation GTF from this genome release were downloaded from release 97 of the Ensembl database (Yates et al. 2020) and used as inputs in subsequent read mapping, annotation, and visualization steps. For each Galaxy tool described below, exact parameters and version numbers used are detailed in Supplementary Table S1. For each sample, reads from across all four NextSeq flow cell lanes were concatenated using the Galaxy Concatenate datasets tail-to-head tool and mapped using RNA STAR (Dobin et al. 2013). Mapped reads were then assigned to exons/genes and tallied using featureCounts (Liao et al. 2014). To enable intersample read count comparisons, count normalization and differential expression analysis was conducted using DESeq2 (Love et al. 2014). Importantly, DESeq2 analysis was performed twice, once on the 12 IP samples and once on the 12 Input samples; see Supplementary Materials and Methods for discussion of this sample separation method.
Outputs from all of the above tools were downloaded from Galaxy for local analysis, computation, and visualization. Custom R scripts were written to generate the scatterplots and hypergeometric test reported here and are available in Supplementary File S3. Scripts in the R programming language (R Core Team 2019) were written and compiled in RStudio (R Studio Team 2018). Additional R packages used in these scripts include ggplot2 (Wickham 2016), ggrepel (Slowikowski 2019), BiocManager (Morgan 2018), and DESeq2 (Love et al. 2014). Analyses were supported by bulk data downloads along with extensive gene-level annotation, sequence information, and references provided by Flybase (Thurmond et al. 2019; Larkin et al. 2021). Principal component analysis (PCA) was conducted by and reported from the above DESeq2 assessment on Galaxy. Mapped reads were visualized in the Integrative Genomics Viewer (Robinson et al. 2011) on the same version of the D. melanogaster genome used above.
Gene-by-gene one-way ANOVAs to identify significantly enriched (i.e., RBP-associated) transcripts
Gene-by-gene ANOVAs and post hoc tests for the 5760 genes identified in the “testable” set, along with bar graphs of IP/Input values, were generated in Prism 8 for Windows 64-bit (GraphPad Software). Custom R and PRISM scripts were written to generate and label the 5760 PRISM data tables, one per testable gene, required for this analysis, and custom R scripts were written to extract and combine the outputs from each test; these scripts are all available in Supplementary File S3. See Results for a summary and below for a further detailed discussion of the statistical testing used to define the testable transcript set and identify significantly enriched (i.e., RBP-associated) transcripts in our RIP-Seq results.
To identify RNA targets of Nab2 and Atx2—that is, RNAs enriched in either Nab2 RIP or Atx2 RIP samples relative to control RIP—directly comparing normalized read counts between RIP samples is insufficient. Differences in RNA expression between samples must be accounted for, as these differences can partially or wholly explain differences in the amount of RNA isolated by IP. We employed a common solution to this problem used in RIP- and ChIP-qPCR (Zhao et al. 2010; Aguilo et al. 2015; Li et al. 2019), scaling normalized RIP reads for each gene in each sample by the corresponding number of normalized Input reads. For clarity, we describe these values as “IP/Input”—they are commonly referred to as “Percent Input” or “% Input.” These IP/Input values could then be compared between samples, further normalizing them to elav-Gal4 alone controls. In this way, RIP fold enrichment, appropriately normalized to library size/composition and gene expression, were calculated for each gene in each sample. To promote the reliability of our analyses and increase our statistical power to detect differences in fold enrichment, we limited further analyses to a testable set of 5760 genes out of the 17,753 total genes annotated in the BDGP6.22 genome. The testable gene set was defined as having detectable expression in all twelve Input samples and an average normalized read count in either Nab2 or Atx2 RIP samples >10. These criteria were based on those used in Lu et al. (2014) and Malmevik et al. (2015). In this defined gene set, differences in fold enrichment were statistically tested using gene-by-gene one-way ANOVAs (Li et al. 2019) in Prism 8 (GraphPad software), applying Dunnett’s post hoc test to calculate significance P-values only for the comparison of each experimental sample to the control sample (Dunnett 1955). In each case, P-values were adjusted to correct for multiple hypothesis testing only within each gene-by-gene ANOVA. This approach identified a small, focused set of statistically significantly enriched RNAs, suggesting that additional corrections across all genes to control type I error (i.e., false positives) were not necessary (Rothman 1990). Due to comparatively low read depth, likely due to incomplete rRNA depletion during library preparation, we suspect that, rather than failing to adequately control type I error, the RBP-associated transcripts we identified through this approach represent a partial census of Nab2 and Atx2 bound RNAs in vivo.
RNA sequencing analysis—sequence motif analyses
Sequence motif analyses were conducted using the MEME Suite of software tools, accessed through the web interface at meme-suite.org (Bailey et al. 2009). For each MEME Suite tool described below, exact parameters and version numbers used are detailed in Supplementary Table S1. Within the MEME Suite, we used MEME itself (Bailey and Elkan 1994) to scan all Nab2-associated transcripts, regardless of their association with Atx2, to (1) identify sequence motifs shared across multiple transcripts and (2) evaluate the frequency and statistical significance of the discovered sequence motifs. Next, FIMO (Grant et al. 2011) was used to quantify the frequency among (1) Nab2-associated transcripts and (2) non-Nab2-associated transcripts of user-provided sequences, specifically (i) a 41 bp A-rich motif identified in Nab2-associated transcripts by MEME, (ii) A12, and (iii) A11G. Non-Nab2-associated transcripts are defined as all 5619 transcripts in the testable set found to not be statistically significantly associated with Nab2 by RIP-Seq. Sequence logos (i.e., visual representations of weighted sequence motifs) were generated by MEME and by WebLogo 3.7.4, available at weblogo.threeplusone.com (Crooks et al. 2004).
Importantly, for any Nab2-associated or non-Nab2-associated transcripts annotated with multiple splice variants, all variant sequences were included as inputs in our motif analyses. This inclusion reflects an inherent limitation of standard shotgun (i.e., short-read) sequencing, as most reads cannot be unambiguously assigned to one splice variant of a given gene, only to given exon(s) encoded by that gene. We therefore chose this inclusion strategy to avoid introducing any bias associated with attempting to call single splice variants for RBP association, and for analytical simplicity. Full sequences of Nab2-associated and non-Nab2-associated transcripts were obtained using the FlyBase Sequence Downloader at flybase.org/download/sequence/batch/(databasereleaseFB2020_04).
Results
Atx2 loss-of-function alleles suppress Nab2 overexpression phenotypes in the adult eye
Previous work established a Gal4-driven Nab2 overexpression system in the Drosophila eye as an effective screening platform to identify potential regulatory partners and targets of Nab2 (Pak et al. 2011; Bienkowski et al. 2017; Lee et al. 2020). This approach uses the Glass Multimer Reporter (GMR) construct (Ellis et al. 1993; Hay et al. 1994) to drive expression of the S. cerevisiae Gal4 transcription factor in fated eye cells (Freeman 1996). In turn, Gal4 binds to Upstream Activating Sequence (UAS) sites within an EP-type P-element (Rorth 1996) inserted upstream of the endogenous Nab2 gene (EP3716) and induces eye-specific overexpression of endogenous Nab2 protein (a genotype hereafter referred to as GMR>Nab2). GMR>Nab2 produces a consistent array of eye morphological defects compared with the GMR-Gal4 transgene control (Pak et al. 2011; Bienkowski et al. 2017; Lee et al. 2020) (Figure 1, A and B). Specifically, this misexpression causes loss of posterior eye pigment, sporadic blackened patches, and disruptions to ommatidial organization lending the surface of the eye a “rough” appearance. Notably, GMR>Nab2-induced pigment loss increases in severity along the anterior-to-posterior axis of the eye, likely because GMR activation occurs behind the morphogenetic furrow, the posterior-to-anterior wave of eye morphogenesis observed in the larval eye disc (Wolff and Ready 1991; Hay et al. 1994). As a result, posterior GMR>Nab2 eye cells experience the longest period of Nab2 overexpression.
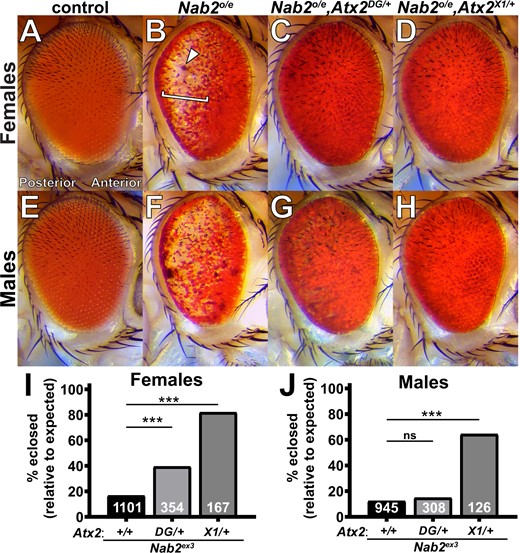
Loss-of-function alleles of Atx2 suppress effects of Nab2 misexpression in female and male Drosophila. Compared with (A) the uniform color and regimented ommatidial eye structure in control females expressing GMR-Gal4 driver alone. (B) Overexpression of endogenous Nab2 with GMR-Gal4 (Nab2°/e) induces posterior pigment loss (bracket), sporadic blackened patches (arrowhead), and ommatidial disordering or “roughness.” Heterozygosity for either of two Atx2 loss-of-function alleles. (C) Atx2DG08112/+ or (D) Atx2X1/+, dominantly suppresses the pigment loss and blackened patch phenotype, with limited impact on roughness. (E–H) These genetic relationships are also observed in eyes in males. (I, J) Nab2ex3 homozygotes lacking endogenous Nab2 show decreased adult viability, as quantified by the percentage of pupal flies eclosing to adulthood out of the amount expected by Mendelian inheritance. (I) In females, both loss-of-function alleles of Atx2 partially rescue this Nab2ex3 reduced viability; (J) in males, only Atx2X1/+ suppresses. Sample sizes (n) are reported in each bar and include all F1 progeny scored, including genetically distinct siblings of the genotype of interest used to calculate % eclosed (relative to expected). Fisher’s exact test (two-sided) was used to assess statistical significance. ns, not significant; ***P< 0.001.
Using the GMR>Nab2 system as a foundation, we previously identified the Drosophila Fragile X Syndrome RBP and neuronal translational regulator Fmr1 as a physical and functional interactor of Nab2 (Bienkowski et al. 2017). An allele of the Ataxin-2 (Atx2) gene, which encodes an RBP that is a regulatory partner of Fmr1 in Drosophila (Sudhakaran et al. 2014), was also detected in this screen as a candidate GMR>Nab2 modifier. To pursue this potential Nab2–Atx2 link, we tested two Atx2 alleles for genetic interactions with GMR>Nab2. The first allele, Atx2DG08112, is caused by the insertion of a 15.6 kb {wHy} P-element near the 5′ end of Atx2 (Huet et al. 2002; Bellen et al. 2004) and is lethal in trans to Df(3R)Exel6174, a deletion that completely removes the Atx2 locus and nearby genes (Parks et al. 2004). Specifically, crossing balanced Atx2DG08112 and Df(3R)Exel6174 alleles produces no trans heterozygotes among the F1 progeny (n = 54). Based on these data, we interpret Atx2DG08112 to be a strong hypomorph. The second Atx2 allele, Atx2X1, is a 1.4 kb imprecise-excision-based deletion that removes the first 22 codons of the Atx2 coding sequence and has been characterized as a null (Satterfield et al. 2002). In part because Nab2 loss induces some sex-specific defects (Jalloh et al. 2021), we analyzed each sex individually. In adult females, heterozygosity for either of these two loss-of-function alleles, Atx2DG08112 (Figure 1C) or Atx2X1 (Figure 1D), dominantly suppresses pigment loss and blackened patches caused by GMR>Nab2 but has limited effect on ommatidial organization or “roughness.” Similar effects of GMR>Nab2 (Figure 1, E and F) and of heterozygosity for either the Atx2DG08112 (Figure 1G) or Atx2X1 (Figure 1H) allele, are observed in males.
Atx2 loss-of-function alleles suppress Nab2 null effects on adult viability and brain morphology
Misexpression of Nab2 induces dramatic phenotypes in domains beyond the eye; homozygosity for the null allele Nab2ex3 causes a dramatic reduction in adult viability (Pak et al. 2011). Thus, to explore whether modifying effects of Atx2 loss-of-function alleles extend to the endogenous Nab2 locus, we analyzed the effect of Atx2 heterozygosity on low adult viability in Nab2ex3 homozygotes. As in the eye, both the Atx2DG08112 and Atx2X1 alleles dominantly suppress the viability defects observed in Nab2ex3 females, elevating adult viability from 17% to 39% and 82%, respectively (Figure 1I). The corresponding effect in males is not as penetrant; only the null Atx2X1 allele dominantly suppresses the viability defect in Nab2ex3 males (Figure 1J). Taken together, these data establish gross similarities in Nab2–Atx2 genetic interactions in females and males. Thus, for practicality we focused further experiments exclusively on female flies, given the more prohibitive impact on male viability of changes in Nab2 expression (Jalloh et al. 2021 and see Materials and Methods).
That Atx2 loss-of-function alleles improve adult viability of Nab2ex3 homozygotes suggests Atx2 and Nab2 coregulate processes or transcripts important for adult development or survival. However, these genetic interactions do not reveal in what cell types or tissues this coregulation may occur. We therefore focused further investigations of Nab2–Atx2 interaction in the brain, given the established and important roles of each protein in brain neurons (Lim and Allada 2013; Sudhakaran et al. 2014; Kelly et al. 2016; Bienkowski et al. 2017). Nab2ex3 homozygous flies develop morphological defects in the axon tracts or lobes of the MB brain structure, a principal olfactory learning and memory center of the insect brain (Heisenberg 2003; Kahsai and Zars 2011; Yagi et al. 2016; Takemura et al. 2017). Specifically, the MBs of surviving Nab2ex3 homozygous null adults exhibit two highly penetrant structural defects: thinning or absence of the dorsally projecting α lobes and overprojection or fusion of the medially projecting β lobes (Kelly et al. 2016). We found that heterozygosity for either Atx2DG08112 or Atx2X1 also causes defects in MB morphology—specifically β lobe fusion—with no apparent effects on α lobe morphology when compared with controls (Figure 2, A–C). Importantly, in the background of Nab2ex3 nulls (Figure 2D), heterozygosity for either Atx2DG08112 (Figure 2E) or Atx2X1 (Figure 2F) suppresses the thinning or absence of α lobes, decreasing the penetrance of this phenotype from 62% of α lobes to 30% or 36%, respectively (Figure 2G). In contrast, neither Atx2 allele as a heterozygote significantly affects the penetrance of β lobe fusion in Nab2ex3 nulls, demonstrating the effect of each mutation is not additive to the effect of Nab2ex3 homozygosity in this context (Figure 2H). A similar α-lobe-specific interaction occurs between alleles of Nab2 and Fmr1 (Bienkowski et al. 2017). Notably, as α and β lobes are composed of tracts of bifurcated axons from single cells (Takemura et al. 2017), this α-lobe-specific suppression by Atx2 alleles demonstrates a Nab2–Atx2 genetic interaction at subcellular resolution. Moreover, that Atx2 loss-of-function alleles suppress defects of a Nab2 null allele implies that Atx2 and Nab2 proteins may coregulate, but in opposing ways, pathways guiding α lobe morphology during development.
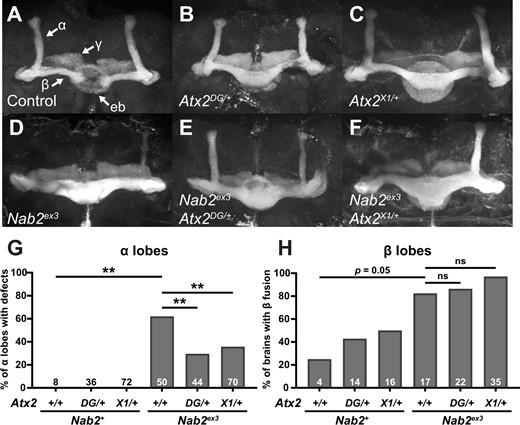
Loss-of-function alleles of Atx2 specifically suppress axon morphology defects in Nab2ex3 MB α, but not β, lobes. (A) In a representative Nab2pex41 control brain, Fasciclin 2 (Fas2)-marked axons from some Kenyon cells of the MB bifurcate and project dorsally into α-lobes and medially into β-lobes. Fas2 also marks γ-lobes and the ellipsoid body (eb) (white arrows). Representative images show heterozygosity for (B) Atx2DG08112/+ or (C) Atx2X1/+ induces overprojection or “fusion” of β-lobes, while (D) homozygosity for the Nab2 null allele Nab2ex3 induces both β-lobe fusion and the thinning or complete absence of α-lobes. Heterozygosity for either (E) Atx2DG08112/+ or (F) Atx2X1/+ in combination with Nab2ex3 partially restores proper α-lobe morphology and (G) significantly suppresses the penetrance of α-lobe defects compared with Nab2ex3 alone. (H) By comparison these Atx2 alleles neither suppress nor enhance the penetrance of β-lobe defects compared with Nab2ex3 alone. Sample sizes (n) are reported in each bar and quantify, for each genotype, the total number of α-lobes scored for defects and the total number of brains scored for β-lobe fusion. Fisher’s exact test (two-tailed) was used to assess statistical significance. ns, not significant; **P≤ 0.01.
Nab2 loss partially suppresses Atx2 circadian phenotypes
Although Nab2 has not previously been identified as a regulator of circadian rhythms, Atx2 loss has been shown to increase circadian period length and decrease the percentage of flies that display rhythmic patterns of behavior (Lim and Allada 2013; Zhang et al. 2013). We reasoned that if Nab2 and Atx2functionally interact to control neuronal morphology, Nab2 loss may modify these Atx2 circadian phenotypes. To test this hypothesis, we determined the free-running circadian period and % rhythmicity of flies where only Atx2, only Nab2, or both Atx2 and Nab2 were reduced in circadian neurons using RNAi (tim-Gal4>UAS-Atx2IR, tim-Gal4>UAS-Nab2IR, and tim-Gal4>UAS-Atx2IR+UAS-Nab2IR, respectively).
During the initial 3-day entrainment period, all genotypes displayed stereotypical patterns of activity at the beginning and end of the light period, demonstrating that they were accurately entrained to a 12-h (12 h) light/dark cycle (Figure 3A). Following the third consecutive day of entrainment, flies were housed in complete darkness for the remainder of the experiment, allowing for the determination of free-running circadian period length (Figure 3B; see also Supplementary Figure S1 and Table S2). As expected, during this 24 h dark period, 100% of w1118 control flies displayed rhythmic behavior patterns, with a free-running circadian period of 23.4 ± 0.33 h, while only 23% of flies lacking the circadian factor period (per01) (Konopka and Benzer 1971) were rhythmic. Flies expressing a Nab2 RNAi line in circadian neurons (tim-Gal4>UAS-Nab2IR) were rhythmic and displayed a free running period of 24.17 ± 0.36 h, which was not significantly different from tim-Gal4 control flies. However, only 3 of 73 flies with Atx2 depleted in circadian cells (tim-Gal4>UAS-Atx2IR) displayed rhythmic patterns of behavior during the 24 h dark period. When Nab2 and Atx2 were both reduced in circadian neurons (tim-Gal4>UAS-Nab2IR + UAS-Atx2IR), the percentage of rhythmic flies increased significantly when compared with flies depleted only for Atx2, consistent with Nab2 loss lessening the severity of circadian defects induced by Atx2 depletion.
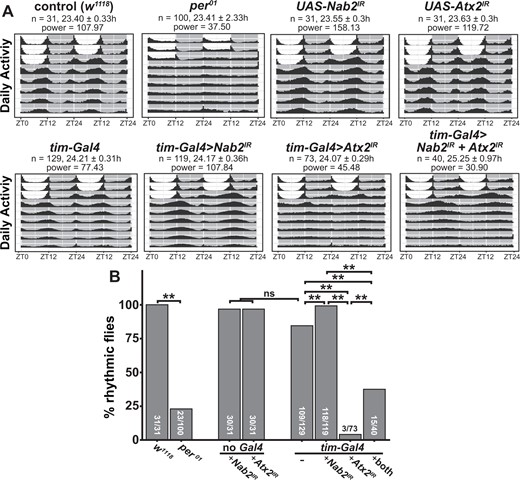
Nab2 loss partially suppresses Atx2-dependent circadian arrhythmicity. (A) Double-plotted actograms showing the activity patterns for male flies of each indicated genotype during 3 days of 12-h light/dark entrainment and 7 days of 24-h darkness. During the 3 days of entrainment, 12-h light periods are shown as white boxes between ZT0 (lights on) and ZT12 (lights off), while 12-h dark periods are shown as gray boxes between ZT12 and ZT24 (which is also ZT0 of the subsequent day). The number of flies (n), calculated circadian period length (hours ± SD), and power of rhythmicity are shown for each genotype. (B) The percentage of rhythmic flies for each genotype as determined by chi-squared periodogram analysis in the Rethomics package (Geissman 2019). For each genotype, the number of rhythmic flies/total number of flies tested is shown. Pairwise Fisher’s exact tests with Bonferroni corrections were performed to assess statistically significant differences between groups (**P < 0.01).
Nab2 and Atx2 primarily localize to distinct compartments in MB neurons
The genetic links between Nab2 and Atx2 could reflect a physical interaction between their encoded proteins (e.g., as shared components of mRNP complexes), as has been observed for both Nab2 and Atx2 with Fmr1 (Sudhakaran et al. 2014; Bienkowski et al. 2017). Alternatively, these genetic links could reflect functional but not physical interactions between Nab2 and Atx2 on common RNAs or neurodevelopmental processes. The latter hypothesis aligns with the localization patterns of each protein—Nab2 localizes primarily to neuronal nuclei with a small fraction in the cytoplasm (Kelly et al. 2016; Bienkowski et al. 2017), while Atx2 concentrates in the neuronal cytoplasm except under certain pathogenic conditions (Lessing and Bonini 2008; Elden et al. 2010). To begin to differentiate between these hypotheses, we evaluated the localization profiles of each protein in MBs in vivo. We expressed both UAS-Nab2-YFP and UAS-Atx2-3xFLAG transgenes in adult MB Kenyon cells using the pan-MB driver OK107-Gal4 (Figure 4A). Similar to human Atx2 in cerebral cortex tissues (Huynh et al. 2003), Drosophila Atx2 primarily localizes to the soma cytoplasm of adult MB Kenyon cells in vivo. In contrast, Nab2 localizes predominantly to the nuclei of these same neurons. A higher magnification view is consistent with only limited overlap between the Atx2-3xFLAG and Nab2-YFP signals (Figure 4B). This pattern of Nab2 and Atx2 localization extends beyond the soma and into the α- and β-lobe axon tracts; Atx2 localizes robustly to the axonal cytoplasm here while Nab2 does not (Supplementary Figure S2). To more rigorously assess the potential for Nab2–Atx2 protein interactions across all cell compartments, we expressed a FLAG-tagged Nab2 transgene (UAS-Nab2-FLAG) (Pak et al. 2011) using the pan-neuronal driver elav-Gal4 (Lin and Goodman 1994) and subjected brain-neuron-enriched head lysates to immunoprecipitation with α-FLAG-conjugated beads to recover Nab2-associated proteins. Probing with specific antibodies confirms that Fmr1 is enriched in Nab2 immunoprecipitates as previously reported (Bienkowski et al. 2017), but reveals only weak enrichment of Atx2 (Figure 4C). These results indicate complexes containing Nab2 and Atx2 may form in neurons but are rare relative to Nab2-Fmr1 complexes. Taken together, these subcellular localization and biochemical data suggest Nab2 and Atx2 do not robustly co-occupy the same RNA or mRNP complexes throughout the post-transcriptional life of an RNA in adult MB neurons. Therefore, we considered the possibility that Nab2–Atx2 genetic interactions instead reflect roles in post-transcriptional control of shared RNA targets at different points in time or different locations in the cell.
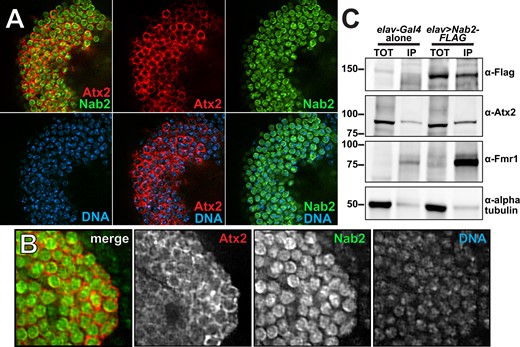
Nab2 and Atx2 proteins primarily localize to different cellular compartments and show limited physical association in brain neurons. To specifically assess protein localization in MB neurons, tagged transgenic copies of Atx2 and Nab2 (Atx2-3xFLAG and Nab2-YFP) were expressed in female brains under the MB-specific OK107-Gal4. Kenyon cell soma (MB cell bodies) are shown for a representative brain (A) with an accompanying high magnificagtion view (B). False-colored panels show fluorescence corresponding to α-FLAG (red, Atx2-3xFLAG), α-GFP (green, Nab2-YFP), Hoechst 33342 (blue, dsDNA), and merges of the indicated channels. Nab2 is localized primarily to the nuclei at steady state based on overlap with Hoechst 33342 signal, and Atx2 localizes primarily in the surrounding cytoplasm. (C) To test for physical association between Nab2 and Atx2 in brain neurons, lysates of adult female heads, either elav-Gal4 alone controls or elav>Nab2-FLAG, were subjected to immunoprecipitation using α-FLAG. For both genotypes, Input samples (TOT) represent 6.25% of assayed lysate and immunoprecipitation (IP) samples represent 25% of total samples eluted from α-FLAG beads. Samples were resolved via gel electrophoresis and analyzed by immunoblotting, probing with antibodies against FLAG, Atx2, Fmr1 (a positive control), or α-tubulin (a negative control). Atx2 associates weakly with Nab2 based on its enrichment in IP samples; this association is less robust than the positive control Nab2-Fmr1 interaction (Bienkowski 2017).
The Nab2 and Atx2 RNA interactomes in brain neurons overlap
Neither Nab2- nor Atx2-associated RNAs have been identified by a high-throughput method in Drosophila—such accounting has been conducted for Atx2 in human cells (Yokoshi et al. 2014) and forNab2 only in S. cerevisiae, not in any metazoan (Kim Guisbert et al. 2005; Batisse et al. 2009; Tuck and Tollervey 2013; Baejen et al. 2014). To test the hypothesis that Nab2 and Atx2 share RNA targets, we identified transcripts stably associated with epitope-tagged versions of each protein in adult brain neurons using an RNA immunoprecipitation-sequencing (RIP-Seq) approach. In this approach, protein products of UAS-Nab2-FLAG or UAS-Atx2-3xFLAG transgenes are robustly expressed under elav-Gal4 control and are efficiently immunoprecipitated from adult head lysates (Figure 5A). Briefly, four biological replicates each of elav-Gal4, elav>Nab2-FLAG, and elav>Atx2-3xFLAG adult female Drosophila heads were lysed and immunoprecipitated with α-FLAG-conjugated beads. Then, RNA from both IP and Input samples was rRNA depleted, reverse transcribed into stranded cDNA libraries, and sequenced. Using the Galaxy web platform through the public server at usegalaxy.org (Afgan et al. 2018), reads were mapped using STAR (Dobin et al. 2013) to the BDGP6.22 release of the D. melanogaster genome (sourced through Ensembl Yates et al. 2020), assigned to exons/genes and tallied using featureCounts (Liao et al. 2014), and normalized for interlibrary count comparisons using DESeq2 (Love et al. 2014). A PCA generated as part of DESeq2 demonstrates the high intergenotype reproducibility among RNA IP (RIP) samples and shows that samples expressing Nab2-FLAG or Atx2-3xFLAG differ more from elav-Gal4 controls than from one another (Figure 5B).
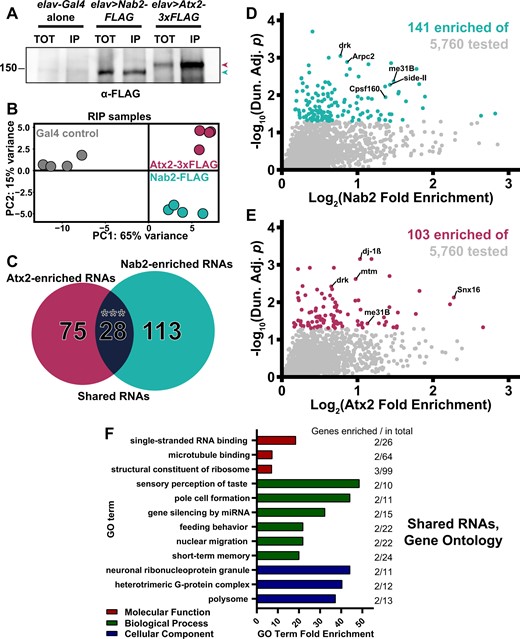
RIP-Seq reveals overlapping sets of transcripts associate with Nab2-FLAG and Atx2-3xFLAG in brain neurons. (A) Lysates from heads of female adult flies expressing either pan-neuronal elav-Gal4 alone as a control, elav>Nab2-FLAG, or elav>Atx2-3xFLAG were subjected to α-FLAG immunoprecipitation and immunoblotting to test IP efficacy. Input samples (TOT) repre-sent ∼6.25% of total assayed lysates and immunoprecipitation samples (IP) represent 25% of total samples eluted from α-FLAG beads. Both epitope-tag samples show robust immunoreactivity to α-FLAG in TOT and IP (arrowheads), indicating effective transgene expression and successful tagged-protein enrichment by IP. (B) PCA of 12 sequenced RIP samples reveals high intragenotype reproducibility. Comparison of principal component 1 (PC1) and principal component 2 (PC2) demonstrates Nab-FLAG (teal) and Atx2-3xFLAG (maroon) samples differ more from “Gal4 alone” controls (gray) than from one another, as predicted. (C) Venn diagram of Nab2-enriched and Atx2-enriched RNAs identified by RIP-Seq, revealing that 28 shared transcripts associate with both RBPs, a significant overlap according to the hypergeometric test (***P < 0.001). Scatter plot of all transcripts within the 5760 of the testable set with positive (D) log2(Nab2 Fold Enrichment) or (E) log2(Atx2 Fold Enrichment) values. Fold enrichment values quantify how effectively a transcript was enriched by IP and are derived by calculating IP/Input (i.e., % input) values for control and epitope-tag samples and setting the average of control values to 1 (i.e., 0 on the logarithmic scale used here). Y-axes display results of significance testing, conducted by gene-by-gene one-way ANOVA, Dunnett’s post hoc test, and within-gene multiple hypothesis testing adjustment (Dun. Adj. P). Statistically significant transcripts (Dun. Adj. P < 0.05) are colored. On each plot, labels identify three transcripts among the “top” (see Results for details) RBP-specific, RBP-associated transcripts and two transcripts (drk, me31B) among the shared RBP-associated transcripts. (F) The independent Molecular Function (red), Biological Process (green), and Cellular Component (blue) Gene Ontology (GO) terms most overrepresented among the shared Nab2- and Atx2-associated transcripts as compared with the entire testable transcript set. GO term independence was determined by “Hierarchical Selection” (see Materials and Methods). The number of GO term members within the shared RBP-associated transcripts and within the entire testable transcript set (Genes enriched/total) are reported to the right of each bar.
To identify Nab2-associated and Atx2-associated RNAs, we calculated % input (IP/Input) enrichment values (Zhao et al. 2010; Aguilo et al. 2015; Li et al. 2019) for each of the 5760 genes in the testable set defined by (1) detectable expression in all 12 Inputs and (2) an average normalized Nab2- or Atx2-IP read count >10 (Lu et al. 2014; Malmevik et al. 2015). Fold enrichment differences were statistically tested by performing gene-by-gene one-way ANOVAs (Li et al. 2019), applying Dunnett’s post hoc test (Dunnett 1955), and calculating adjusted P-values corrected for multiple hypothesis testing within each gene-by-gene ANOVA (values hereafter referred to as Dun. Adj. P; see Materials and Methods for more details). Using this approach, we identify 141 and 103 RNAs significantly enriched in α-FLAG IPs of elav>Nab2-FLAG and elav>Atx2-3xFLAG female heads, respectively (Supplementary Table S3 and Figure S3). The size and focus of these sets of statistically significantly enriched RNAs suggests type I (i.e., false positive) error is sufficiently controlled and additional corrections between genes are not necessary (Rothman 1990). Comparing the Nab2- and Atx2-IP groups strongly supports our hypothesis, revealing 28 transcripts shared between Nab2- and Atx2-associated Drosophila neuronal RNAs (Figure 5C). This overlap is highly significant according to the hypergeometric test—it is extremely unlikely to occur by random selection from the total tested gene set. The full list of transcripts associated with both Nab2 and Atx2 (Table 1) includes multiple mRNAs that encode proteins with functions in neuronal domains in which Nab2 and Atx2 genetically interact, raising the possibility that coregulation of these RNAs by Nab2 and Atx2 partially explains these Nab2–Atx2 genetic links. These shared transcripts include drk (downstream of receptor kinase), me31B (maternal expression at 31B), sm (smooth), and stai (stathmin). The protein encoded by drk is a receptor tyrosine kinase (RTK) adaptor that regulates growth and development by binding activated RTKs, such as sevenless in R7 retinal cells (Almudi et al. 2010), and contributes to, among other processes, cell survival in the eye (Schoenherr et al. 2012) and olfactory learning and memory in the MB (Moressis et al. 2009). The protein encoded by me31B is a DEAD-box RNA helicase expressed in many cellular contexts, including the MB Kenyon cells (Hillebrand et al. 2010) and the oocyte (Nakamura et al. 2001), that physically associates with Atx2 (Lee et al. 2017) and serves as a central player in miRNA-mediated translational repression (Barbee et al. 2006) and assembly of some RNP granules (Eulalio et al. 2007). Finally, the proteins encoded by sm and stai are, respectively, an hnRNP linked to defects in axon termination (Layalle et al. 2005) and an axonal tubulin-binding protein linked to natural variation in the size of MB α and β lobes (Lachkar et al. 2010; Duncan et al. 2013; Zwarts et al. 2015).
Identities of the 28 transcripts overlapping between the Nab2 and Atx2 RNA interactomes
| Shared Nab2- and Atx2-associated transcripts | |||
|---|---|---|---|
| AGO2 | Drk | me31B | shi |
| Apolpp | Gαo | Msp300 | sm |
| CG31221 | Gat | mtd | snoRNA: Or-aca5 |
| CG42540 | Gγ30A | Rbp6 | snoRNA: Or-CD2 |
| CG4360 | Gp150 | RpL37A | snoRNA: Ψ18S-1854b |
| CG6675 | HmgZ | RpS27A | stai |
| CG9813 | l(3)80Fga | RpS29 | Ulp1 |
| Shared Nab2- and Atx2-associated transcripts | |||
|---|---|---|---|
| AGO2 | Drk | me31B | shi |
| Apolpp | Gαo | Msp300 | sm |
| CG31221 | Gat | mtd | snoRNA: Or-aca5 |
| CG42540 | Gγ30A | Rbp6 | snoRNA: Or-CD2 |
| CG4360 | Gp150 | RpL37A | snoRNA: Ψ18S-1854b |
| CG6675 | HmgZ | RpS27A | stai |
| CG9813 | l(3)80Fga | RpS29 | Ulp1 |
For all 5760 genes in the RIP-Seq testable set, control-normalized IP/Input enrichment values were calculated followed by gene-by-gene one-way ANOVAs, Dunnett’s post hoc tests, and within-gene multiple hypothesis testing adjustment (Dun. Adj. P). All transcripts statistically significantly (Dun. Adj. P < 0.05) enriched in both Nab2- and Atx2-associated transcripts sets are listed here.
Symbol updated from CG40178 to current BDGP6.37 nomenclature.
Identities of the 28 transcripts overlapping between the Nab2 and Atx2 RNA interactomes
| Shared Nab2- and Atx2-associated transcripts | |||
|---|---|---|---|
| AGO2 | Drk | me31B | shi |
| Apolpp | Gαo | Msp300 | sm |
| CG31221 | Gat | mtd | snoRNA: Or-aca5 |
| CG42540 | Gγ30A | Rbp6 | snoRNA: Or-CD2 |
| CG4360 | Gp150 | RpL37A | snoRNA: Ψ18S-1854b |
| CG6675 | HmgZ | RpS27A | stai |
| CG9813 | l(3)80Fga | RpS29 | Ulp1 |
| Shared Nab2- and Atx2-associated transcripts | |||
|---|---|---|---|
| AGO2 | Drk | me31B | shi |
| Apolpp | Gαo | Msp300 | sm |
| CG31221 | Gat | mtd | snoRNA: Or-aca5 |
| CG42540 | Gγ30A | Rbp6 | snoRNA: Or-CD2 |
| CG4360 | Gp150 | RpL37A | snoRNA: Ψ18S-1854b |
| CG6675 | HmgZ | RpS27A | stai |
| CG9813 | l(3)80Fga | RpS29 | Ulp1 |
For all 5760 genes in the RIP-Seq testable set, control-normalized IP/Input enrichment values were calculated followed by gene-by-gene one-way ANOVAs, Dunnett’s post hoc tests, and within-gene multiple hypothesis testing adjustment (Dun. Adj. P). All transcripts statistically significantly (Dun. Adj. P < 0.05) enriched in both Nab2- and Atx2-associated transcripts sets are listed here.
Symbol updated from CG40178 to current BDGP6.37 nomenclature.
The 28 shared transcripts represent approximately 20% and 24% of the total transcripts identified as Nab2- and Atx2-associated, respectively, underscoring that these proteins also associate with RNA sets independent from one another. From these independent sets, we defined the top Nab2-specific and Atx2-specific associated transcripts as the top 20 most significantly associated transcripts (by Dun. Adj. P) and top 20 most strongly enriched transcripts (by IP/Input) in each set. As with shared RNAs, multiple RBP-specific RNAs with links to Nab2 or Atx2 functions or mutant phenotypes are identified among these top transcripts, raising the possibility that regulation of these RNAs by Nab2 or Atx2 partially explains the mechanism of action of these RBPs (Figure 5, D and E). For example, the top Nab2-specific associated RNAs include Arpc2 (Actin-related protein 2/3 complex, subunit 2), side-II (sidestep II), and Cpsf160 (Cleavage and polyadenylation specificity factor 160). These transcripts, respectively, encode proteins with proposed functions in neuronal growth cone advance (Yang et al. 2012), synapse formation between certain neuronal subtypes (Tan et al. 2015), and mRNA poly(A)-tail formation based on orthology to mammalian Cpsf1 (Mandel et al. 2008). The top Atx2-specific associated RNAs include dj-1β, mtm (myotubularin), and Snx16 (Sorting nexin 16). These transcripts, respectively, encode proteins with proposed functions in ATP synthesis and motor neuron synaptic transmission (Hao et al. 2010; Oswald et al. 2018), endosomal trafficking regulation via phosphatase activity (Velichkova et al. 2010; Jean et al. 2012), and neuromuscular junction synaptic growth (Rodal et al. 2011).
Gene Ontology terms enriched in Nab2 and Atx2 RNA interactomes emphasize additional RBP-associated transcripts
Evaluating Nab2- and Atx2-associated RNAs individually provides valuable but incomplete insight, allowing larger trends to be missed. Thus, to complement these analyses we evaluated the shared and specific Nab2- and Atx2-associated transcript sets by subjecting each gene list to PANTHER Gene Ontology (GO) analysis, revealing the identities and members of enriched GO terms in each transcript set (Ashburner et al. 2000; Mi et al. 2019; The Gene Ontology Consortium 2019). GO term enrichment was calculated by comparing term abundance between these lists and the testable set of 5760 head-enriched genes rather than the entire genome. In this way, these analyses did not identify GO terms as enriched simply because of their overrepresentation in Drosophila heads. Among shared Nab2- and Atx2-associated RNAs, we identified overrepresented GO terms and RBP-associated transcripts within them that highlight crucial functions and processes Nab2 and Atx2 may coregulate (Figure 5F). Among these GO terms are “microtubule binding,” which includes apolpp (apolipophorin) and shi (shibire); “sensory perception of taste,” which includes Gαo and Gγ30A; “gene silencing by miRNA,” which includes AGO2 (Argonaute 2) and me31B; and “short-term memory,” which includes shi and drk. Survey of the associated RNAs specific to either RBP reveals overrepresented GO terms and transcripts within them which may mediate processes Nab2 and Atx2 regulate independently of one another, including, respectively, the GO terms “exosomal secretion,” which includes Rab35 and Rab7; and “regulation of ATP metabolic process,” which includes Dg (Dystroglycan) and dj-1β (Supplementary Figure S4).
To combine and summarize the individual transcript and GO analyses, we highlight groups of seven transcripts found within the shared (Figure 6A) and RBP-specific (Figure 6, B and C) associated transcript sets. These highlights were selected from the combined set of transcripts (1) demonstrating a fold enrichment (IP/Input) >1.5, and/or (2) included in the most overrepresented GO terms (fully defined in Supplementary Table S4). Beyond transcripts already described, this summary includes the shared transcript HmgZ (HMG protein Z), Nab2-specific transcripts fwe (flower) and SLC22A (SLC22A family member), and Atx2-specific transcripts tea (telomere ends associated) and Xpc (Xeroderma pigmentosum, complementation group C). A group of functionally diverse transcripts in the testable set that did not associate with either RBP is shown for comparison and as evidence of the specificity of the RIP-Seq assay (Figure 6D).
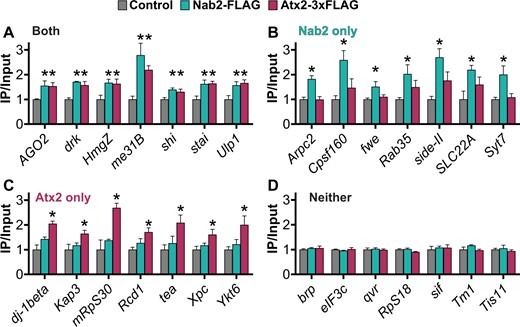
Potential functionally important RNA targets of Nab2 and Atx2 identified by combining individual transcript and GO analyses of RIP-Seq results. For transcripts that associate with both Nab2 and Atx2, Nab2 only, Atx2 only, or neither RBP by RIP-Seq, seven transcripts of particular functional interest are presented as a summary of each category. (A–C) These transcripts met one or both of two criteria: (1) inclusion in an associated overrepresented GO term (2) an IP/Input (i.e., fold enrichment) value >1.5. Given the functions of proteins encoded by these transcripts, these selections represent potential phenotypically important targets of post-transcriptional regulation by Nab2 and Atx2. (D) Negative control transcripts encode a functionally diverse set of proteins that do not associate with Nab2 or Atx2. Error bars represent standard errors of the mean (SEM). Gene-by-gene one-way ANOVA, Dunnett’s post hoc test, and within-gene multiple hypothesis testing adjustment (Dun. Adj. P) was used to assess statistical significance. *Dun. Adj. P < 0.05.
Polyadenosine sequence motifs are enriched in Nab2-associated RNAs
The diversity of RNAs that do not associate with Nab2 and Atx2 in the RIP assay supports a key finding—both RBPs exhibit specific RNA-association patterns within brain neurons. This observation is not surprising for Atx2 given, for example, the sequence specificity of its human homolog in HEK293T cells (Yokoshi et al. 2014), but it represents a valuable insight for Nab2. The extent of the metazoan Nab2/ZC3H14 RNA target pool has been an enduring question (Rha et al. 2017a), in part given the breadth of the S. cerevisiae Nab2 target pool (Batisse et al. 2009; Tuck and Tollervey 2013). Moreover, the ability of Nab2/ZC3H14 across eukaryotes to bind polyadenosine RNA in vitro (Kelly et al. 2007; Pak et al. 2011) raises the possibility for Nab2/ZC3H14 to bind very broadly to mRNAs via their poly(A) tails in vivo. We found a relatively focused set of RNAs coprecipitate with Nab2-FLAG from fly brain neurons, indicating Nab2 may indeed exhibit greater specificity in Drosophila than would be observed if the protein bound broadly to all or most polyadenylated transcripts via their poly(A) tails.
Thus, we sought to determine what additional RNA sequence features may drive the association of Nab2 with its target transcripts if not the presence of a poly(A) tail alone. We used MEME (Bailey and Elkan 1994) to scan all Nab2-associated transcripts to identify shared sequence motifs that may represent Nab2 binding sites and partially explain Nab2 specificity. Strikingly, this analysis identifies a 41 bp long, internal-A-rich stretch among the top 10 motifs (6–50 bp in length) shared among Nab2-associated transcripts. Importantly, each of these 10 sequence motifs are shared across overlapping sets of many but not all Nab2-associated RNAs. This identification of an A-rich stretch enriched in Nab2 associated transcripts is compelling as extensive biochemistry has shown that the zinc fingers present in Nab2 bind with high affinity and specificity to polyadenosine RNA tracts (Kelly et al. 2007, 2010; Aibara et al. 2017). Furthermore, an unbiased approach defined the sequence A11G as a Nab2-binding motif in budding yeast (Kim Guisbert et al. 2005). Thus, the results of our MEME motif analysis agree with extensive prior biochemical analysis of Nab2.
Using FIMO (Grant et al. 2011), another part of the MEME Suite (Bailey et al. 2009), we quantified the frequency of close and exact matches to the consensus version of this A-rich motif among Nab2-associated RNAs. Such occurrences are significantly more common in Nab2-associated transcripts compared with non-Nab2-associated transcripts, respectively, appearing once every 135 bases and once every 845 bases on average, a 6.3-fold enrichment (Figure 7A). The high frequency of this motif in Nab2-associated transcripts is consistent with data from S. cerevisiae that Nab2 does not associate with RNAs exclusively through the poly(A) tail and also binds to upstream UTRs and coding sequences, likely through other A-rich sequences (Kim Guisbert et al. 2005; Gonzalez-Aguilera et al. 2011; Tuck and Tollervey 2013; Baejen et al. 2014; Aibara et al. 2017). Importantly, that this A-rich motif is enriched in, but not exclusive to, Nab2-associated RNAs is consistent with results for other RBPs. Linear sequence motifs alone are generally insufficient to explain RBP specificity (Dominguez et al. 2018) and RBPs do not generally occupy all of their available binding motifs throughout the transcriptome (Li et al. 2010; Taliaferro et al. 2016).
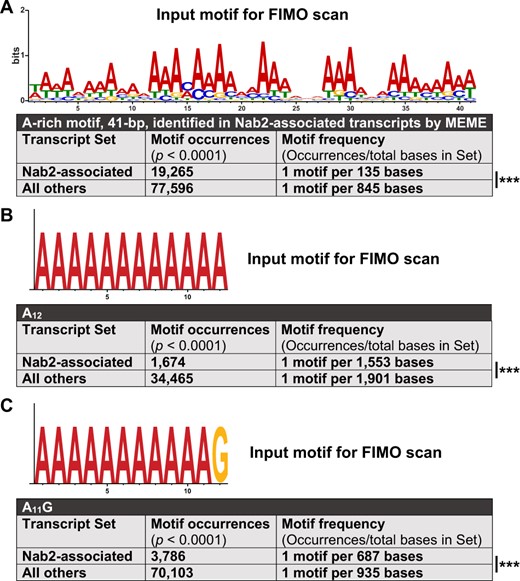
A broad A-rich motif and two established Nab2 binding motifs are enriched in Nab2-associated RNAs. Output from transcript set scans by FIMO, which quantifies the occurrences motifs identical or highly similar to an input motif. Two transcript sets were scanned in each analysis: (1) all transcripts encoded by Nab2-associated gene models and (2) all transcripts for non-Nab2-associated gene models in the RIP-Seq testable set. (A) A 41-bp A-rich motif, identified by MEME as one of the first ten 6–50 bp motifs within Nab2-associated transcripts, was used as input for FIMO. (B) A canonical Nab2-binding motif from S. cerevisiae, A11G, was used as FIMO input. (C) A homopolymeric A12 motif was used as FIMO input. In all three cases, particularly in (A), the scanned motif is significantly enriched in the Nab2-associated transcript set compared with the non-Nab2-associated set. However, none of the three input motifs are exclusive or nearly exclusive to Nab2-associated transcripts—each is still notably abundant in the complete dataset. Statistical significance was assessed using the chi-square test (two-sided). ***P < 0.001.
As a complement to these analyses, we used FIMO to scan Nab2-associated RNAs for the presence of the smallest canonical binding motifs sufficient for Nab2 association in S. cerevisiae, A12 and A11G (Kim Guisbert et al. 2005; Aibara et al. 2017). This approach reveals that in Drosophila brain neurons A12 and A11G sites are significantly but moderately more common in Nab2-associated transcripts compared with non-Nab2-associated transcripts. These A12 and A11G sites appear, respectively, once every 1553 and 687 bases on average among Nab2-associated transcripts and once every 1901 and 935 bases on average among non-Nab2-assoicated transcripts, a 1.2- and 1.4-fold enrichment (Figure 7, B and C). Taken together, the findings that Nab2 associates with a specific subset of all RNAs with poly(A) tails, and that these three A-rich motifs are not exclusive to Nab2-associated RNAs, argues that the polyadenosine sequence affinity of Nab2 alone is insufficient to explain Nab2–RNA association specificity in Drosophila brain neurons. Other mechanisms must also contribute to Nab2 target choice, such as RNA secondary structure, protein-protein interactions, subnuclear localization, and binding site competition. That said, the significant enrichment of 41 bp A-rich, A12, and A11G motifs in Nab2-associated RNAs suggests Nab2–RNA association is partially mediated through these genetically encoded RNA sequence motifs as well as or instead of through the poly(A) tail.
Alleles of genes encoding Nab2-associated mRNAs modify GMR>Nab2
To test the in vivo biological relevance of Nab2-mRNA physical associations identified in the RIP-Seq, we obtained loss-of-function alleles corresponding to 10 Nab2-associated mRNAs (drk, stai, Arpc2, Bsg, SLC22A, Gαo, mtd, me31B, HmgZ, and sm) and assessed each for effects in the GMR>Nab2 eye model. Among these 10 alleles, we detected eight modifiers: seven suppressors, Arpc2KG04658, Bsgk13638, GαoKO, sm1, staiKO, mtd4, HmgZEY16326 (inserted in the HmgZ 5′ UTR intron in the opposite direction of transcription), and one enhancer, drk0A (Figure 8). GαoKO and Df(3L)Exel6137 (a deficiency encompassing SLC22A and the neighboring genes CG14562, PolH, and mub) moderately suppress GMR>Nab2, while me31Bk06607 had little effect. Notably, mRNAs of two of the suppressors, Bsg and mtd, show specific intron-retention defects in high-throughput RNA sequencing analysis of Nab2 mutant adult heads (Jalloh et al. 2021), while a third, stai, shows reduced frequency of mRNA species that initiate from a 5′ exon 1 (stai-RC and RE), which is predicted to affect the translation start-site and 5′-UTR structure (Supplementary Figure S5). Overall, these data indicate the Nab2-associated mRNAs identified by neuronal Nab2-FLAG RIP-Seq are enriched for mRNAs regulated by Nab2 and imply potential regulatory roles for Nab2 on the Bsg, stai, and mtd RNAs in neurons.
![Enrichment for Nab2 genetic interactors among 10 candidate loci identified by Nab2 RIP-Seq. White light images of adult female eyes from (A) control (Oregon R), or (B) GMR>Nab2 alone (+/+) or (C–L) in combination with each of the ten indicated alleles. Eight of these ten alleles dominantly modify the “small, rough eye” phenotype characteristic of GMR>Nab2 (i.e., Nab2°/e). Seven of these are suppressors: Arpc2, HmgZ, sm, stai, and mtd alleles suppress strongly (C, G, J–L), with restoration of pigmentation, size, and some ommatidial patterning in the anterior portion of the eye, while the Bsg and Gαo only incompletely restore pigmentation (D, F). The drk allele behaves as a dominant enhancer (E) which reduces eye size, increases pigment loss, and leads to the appearance of black spots. The final two alleles, me31B and SLC22A [in the Df(3L)Exel6137 deficiency] (H, I) have no effect, or a very mild effect, on GMR>Nab2 phenotypes.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/genetics/220/1/10.1093_genetics_iyab175/2/m_iyab175f8.jpeg?Expires=1716320317&Signature=dVxHgqK-N4C1uZ8z-y-oy9u3fKPcRbsmxYzy4T16ebLJN4rJvSDsOj12zUg~EHZ1n0~L72mRMUWG3gm3nEUfvy4wJuh1LvvgDaPDPZkk5kJD6hYocjPFC2fGJXKLpps00RDt5cEeQrqVaF51l6UMnjl0UEWzWiLlmPHOX2uF5Hg0QnLReAySu1ispjqvXfxtl6n6merMTYZAzn-xuY6Y6CpHV8lxmDL5wF30XakmNHZ6vbY-BB2MifjxApK5BgxyU7u7QBsZJOrrf0kDYfUBjjQ8UUhDvfrVYPATnYyhp8Ke9zcFDcT50BvPC-7jA6WIcclZA0uHBUDmuWIqMfFnvA__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Enrichment for Nab2 genetic interactors among 10 candidate loci identified by Nab2 RIP-Seq. White light images of adult female eyes from (A) control (Oregon R), or (B) GMR>Nab2 alone (+/+) or (C–L) in combination with each of the ten indicated alleles. Eight of these ten alleles dominantly modify the “small, rough eye” phenotype characteristic of GMR>Nab2 (i.e., Nab2°/e). Seven of these are suppressors: Arpc2, HmgZ, sm, stai, and mtd alleles suppress strongly (C, G, J–L), with restoration of pigmentation, size, and some ommatidial patterning in the anterior portion of the eye, while the Bsg and Gαo only incompletely restore pigmentation (D, F). The drk allele behaves as a dominant enhancer (E) which reduces eye size, increases pigment loss, and leads to the appearance of black spots. The final two alleles, me31B and SLC22A [in the Df(3L)Exel6137 deficiency] (H, I) have no effect, or a very mild effect, on GMR>Nab2 phenotypes.
Discussion
Mutation of either ZC3H14 or ATXN2 gives rise to human disease, and the Nab2 and Atx2 RBPs encoded by their Drosophila orthologs are linked by a shared association with Fmr1 (Sudhakaran et al. 2014; Bienkowski et al. 2017). Here, we show that Nab2 and Atx2 interact in multiple contexts in Drosophila, specifically in fated eye cells, adult viability, MB neuronal morphology, and circadian behavior. Notably, these interactions are dose-sensitive, as heterozygosity for Atx2 loss-of-function alleles is sufficient to suppress Nab2 null phenotypes in adult viability and MB morphology. That is, loss of Nab2 may sensitize these domains to reduced Atx2 activity, suggesting these RBPs regulate some common processes. We find that these Nab2–Atx2 interactions are likely not explained by extended, simultaneous co-occupancy of Nab2 and Atx2 in common RNP complexes on shared RNA transcripts. Each protein is mainly concentrated in distinct subcellular compartments in adult MB neurons in vivo with a small amount of potential overlap at the nuclear periphery, and Nab2 and Atx2 only weakly associate by co-IP from brain neurons. To explore alternative models (e.g., sequential regulation of shared RNA transcripts), we have carried out the first high-throughput identification of Nab2- and Atx2-associated RNAs in Drosophila. We find these proteins associate with overlapping sets of transcripts in Drosophila neurons, consistent with their shared and distinct functions and supporting the model of sequential regulation. Finally, to assess the in vivo relevance of these physical interactions, we tested and confirmed that alleles corresponding to a subset of Nab2-associated mRNAs act as dose-sensitive modifiers of Nab2 overexpression phenotypes in the developing eye. In sum, identification of these protein-transcript associations and corresponding genetic interactions promises further insight into the functions shared between and unique to each RBP. The identification of Drosophila Nab2-associated RNAs also begins to address longstanding questions about Nab2 function and the particular sensitivity of neurons to Nab2 loss, revealing that Nab2 associates with a specific subset of polyadenylated RNAs in vivo despite the theoretical potential to bind across all polyadenylated transcripts suggested by its high polyadenosine affinity in vitro (Pak et al. 2011).
A model of opposing regulatory roles for Nab2 and Atx2
We present evidence that Nab2 and Atx2 share associated RNAs in Drosophila neurons (Figures 5 and 6), that Atx2 loss-of-function alleles suppress phenotypes of Nab2 loss (Figures 1 and 2), and that reduction in Nab2 protein level suppresses circadian phenotypes of reduction in Atx2 protein level (Figure 3). Taken together, these findings imply that, at least for transcripts crucial for adult survival, MB α-lobe morphology, and circadian behavior, Nab2 and Atx2 exert opposing regulatory roles on a set of shared associated RNAs. This opposing role possibility aligns with some of the known functions of each protein. Namely, in S. cerevisiae Nab2 contributes to proper nuclear processing events including protection from enzymatic degradation, poly(A) tail length control, proper splicing, and transcriptional termination while also facilitating poly(A) RNA export from the nucleus (Green et al. 2002; Hector et al. 2002; Kelly et al. 2010; Schmid et al. 2015; Soucek et al. 2016; Alpert et al. 2020). If Drosophila Nab2 also performs some, or all, of these nuclear processing roles on associated RNAs, then Nab2 binding should contribute to transcript stability, nuclear export, and ultimately protein expression. Atx2, in contrast, is a key regulator of translational efficiency in the cytoplasm, suppressing the translation of some target RNAs and activating the translation of others (reviewed in Lee et al. 2018). As our data suggest Nab2 and Atx2 act in functional opposition on a shared transcript set, we propose Atx2 primarily functions as a translational inhibitor rather than activator on shared Nab2- and Atx2-associated RNAs. In this model (Figure 9), Nab2 and Atx2 would act in temporal and spatial sequence to balance protein expression from their shared associated RNAs in neurons, with Nab2 promoting proper nuclear RNA processing, stability, and export and Atx2 inhibiting RNA translation, respectively.
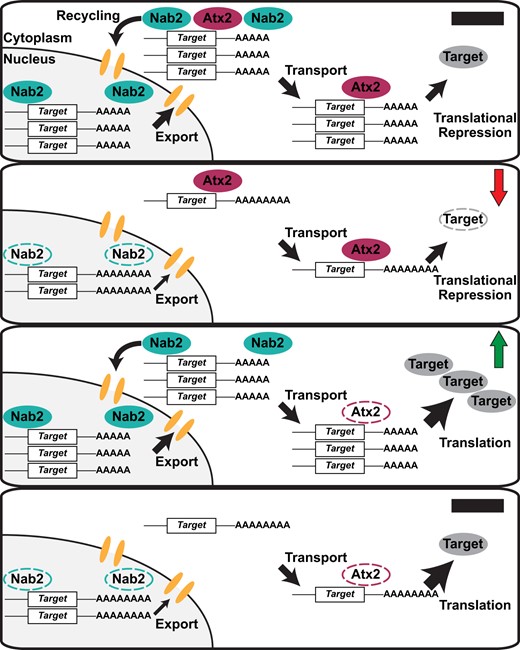
A model of opposing regulatory roles for Nab2 and Atx2 on shared associated RNA transcripts. (Top panel) In the wild-type condition, Nab2 protects transcripts from degradation, limits poly(A) tail length, and contributes to target RNA export from the nucleus, shuttling with its associated transcripts into the cytoplasm. Nab2 and Atx2 may co-occupy the same transcripts briefly or occasionally during nuclear-cytoplasmic mRNP remodeling and prior to Nab2 recycling into the nucleus. Atx2 accompanies target transcripts to their destinations (e.g., synaptic terminals) and contributes to miRNA-mediated translational repression, which is released under certain conditions (e.g., synaptic activity), ultimately contributing to regulated production of the encoded protein. (Second panel) In Nab2ex3 nulls, target mRNAs are less stable, exhibit longer poly(A) tails, and are exported less efficiently from the nucleus. As a result, less mRNA reaches its appropriate destination, resulting in a decrease in steady-state levels of encoded protein (red arrow). (Third panel) In Atx2 loss-of-function heterozygotes (i.e., Atx2DG08112/+ or Atx2X1/+), less Atx2 protein is expressed and available to repress target translation, resulting in less responsive, higher steady-state levels of encoded protein (green arrow). (Bottom panel) Effects of the complete loss of Nab2 in Nab2ex3 and the decrease of functional Atx2 in Atx2 loss-of-function heterozygotes balance one another. While nuclear target mRNA is less stable and less is exported from the nucleus successfully, these RNAs are also under less strict translational control in partial absence of Atx2, ultimately resulting in balanced effects on encoded protein levels.
This model of sequential temporal and spatial regulation aligns with evidence that Nab2 and Atx2 primarily localize to different subcellular compartments in adult MBs at steady state, with potential overlap at the nuclear periphery, and exhibit a low level of coprecipitation from brain neurons (Figure 4). Potential explanations for the combination of localization profiles and limited physical association between Nab2 and Atx2 are found in proposals that S. cerevisiae Nab2 shuttles out of the nucleus with bound RNAs during export before releasing these RNAs and returning to the nucleus (Aitchison et al. 1996; Lee and Aitchison 1999; Duncan et al. 2000). Thus, Nab2 and Atx2 may physically share associated RNAs briefly at the nuclear periphery if neuronal Drosophila Nab2 similarly shuttles and both RBPs are present during the nuclear-cytoplasmic handoff of mRNP remodeling that follows mRNA export from the nucleus (reviewed in Muller-McNicoll and Neugebauer 2013; Chen and Shyu 2014). Functional and physical links between Nab2 and an RBP with a prominent cytoplasmic localization pattern like Atx2 have been observed previously, specifically with Fmr1 (Bienkowski et al. 2017). However, the physical associations observed between Fmr1 and Nab2 are more robust than that observed between Atx2 and Nab2 in the present study (Figure 4C)—this distinction may be partially explained by the different localization patterns of Atx2 and Fmr1. Atx2 is exclusively cytoplasmic in neurons except under certain pathogenic conditions (Huynh et al. 2003; Lessing and Bonini 2008; Elden et al. 2010), while Fmr1 shuttles between the two compartments, associating with at least some of its target RNAs in the nucleus (Tamanini et al. 1999; Kim et al. 2009). Thus, Nab2 and Fmr1 may theoretically co-occupy and coregulate shared transcripts in both cellular compartments while Nab2 and Atx2 sequentially regulate shared transcripts exchanged during a nuclear-cytoplasmic handoff, representing two distinct modes of functional interaction between Nab2 and a cytoplasmic RBP.
This model provides a firm foundation and raises many readily testable hypotheses to be explored in future research. The model predicts that for shared Nab2- and Atx2-associated RNAs, loss of Nab2 decreases transcript stability, impedes proper nuclear processing events including poly(A) tail length control, and impairs poly(A) RNA export from the nucleus, ultimately leading to decreases in protein product. Conversely, we predict partial loss of Atx2 releases translational inhibition on these shared transcripts and induces increases in protein product. Finally, loss of both proteins would balance these effects, resulting in steady-state levels of protein product more similar to the wild-type condition. With the identity of Nab2- and Atx2-associated RNAs in hand, future research is enabled to test these predictions.
Prominent Nab2- and Atx2-associated transcripts provide links to brain development and function
We define the prominent shared and RBP-specific associated transcripts as those annotated within overrepresented GO terms (Figure 5F; Supplementary Figure S4) and/or passing a 1.5-fold enrichment threshold (examples in Figure 6). The identities and functional roles of these prominent transcripts provide potential mechanistic explanations for biological effects of Nab2 and Atx2 loss in the nervous system. For example, the effects of Nab2 and Atx2 loss on MB morphology may be mediated in part through altered regulation of Nab2- and Atx2-associated mRNAs such as sm, which encodes an hnRNP involved in axon guidance, Bsg, which encodes a IgG-family glycoprotein homologous to human Neuroplastin that interacts with integrin to uphold the structure of the glia-extracellular matrix in the Drosophila brain, and stai, which encodes a tubulin-binding protein linked to axonal morphology and development (Layalle et al. 2005; Lachkar et al. 2010; Graf et al. 2011; Duncan et al. 2013; Zwarts et al. 2015; Hunter et al. 2020). Similarly, effects of Nab2 and Atx2 alleles on memory (Sudhakaran et al. 2014; Kelly et al. 2016) may be due in part to altered regulation of shared transcripts drk, shi, Gαo, and me31B, all of which encode proteins with roles in memory formation or retrieval (Dubnau et al. 2001; Ferris et al. 2006; Moressis et al. 2009; Sudhakaran et al. 2014). Both Nab2 and Atx2 may be involved in the RNAi pathway at multiple levels, regulating me31B RNA in neurons in addition to associating, in the case of Atx2, with Me31B protein (Lee et al. 2017; Bakthavachalu et al. 2018). The suppression of GMR>Nab2 by Atx2 alleles in the eye may be explained in part by the shared association of Nab2 and Atx2 with HmgZ (HMG protein Z) mRNA, which encodes a chromatin remodeler linked to survival of R7 retinal photoreceptor neurons (Kanuka et al. 2005; Ragab et al. 2006). Similarly, neurodevelopmental phenotypes in Nab2ex3 null brains could stem in part from deregulation of Nab2-specific associated mRNAs such as mtd, which encodes a transcriptional coactivator homologous to human OXR1, loss of which causes brain atrophy (Wang et al. 2019), and Arpc2, which encodes a component of the neuronal growth cone advance-regulating Arp2/3 complex (Hudson and Cooley 2002; Yang et al. 2012). While these mechanistic links are speculative, the strong genetic interactions we detect between GMR>Nab2 and alleles of Bsg, stai, mtd, Arpc2, sm, HmgZ, and drk support a model in which physical association of these RNAs with Nab2 is paralleled by a role for Nab2 in post-transcriptional regulation of these RNAs in neurons.
Moreover, among the associated RNAs specific to each RBP, we found only Nab2 associated with fwe (flower), side-II, and SLC22A RNA, connections which may further explain the role of Nab2 in guiding MB morphology and regulating learning and memory. These transcripts, respectively, encode a transmembrane mediator of neuronal culling in development (Merino et al. 2013), an immunoglobulin superfamily member potentially contributing to axon guidance and synapse formation in the optic lobe (Tan et al. 2015), and a transmembrane acetylcholine transporter localized to MB dendrites and involved in suppressing memory formation (Gai et al. 2016). On the other hand, the association of Atx2 with Atx2-specific RNAs Xpc and tea, which, respectively, encode players in the fundamental cellular processes of DNA repair and telomere protection (Henning et al. 1994; Goosen 2010; Zhang et al. 2016), may partially explain why Atx2 genomic loss, unlike Nab2 genomic loss, is larval lethal (Satterfield et al. 2002). In summary, defining the potential functional impact of Nab2- and Atx2–RNA associations like these will provide critical insight into the roles of Nab2 and Atx2 in neurodevelopment and Drosophila disease models.
Nab2 associates with a more specific set of RNAs in metazoans than in S. cerevisiae
The degree of RNA association specificity metazoan Nab2/ZC3H14 exhibits has been a longstanding question, in part because competing answers are suggested by the functional similarities and differences between metazoan Nab2/ZC3H14 and the S. cerevisiae Nab2 ortholog. In S. cerevisiae, Nab2 is essential for viability (Anderson et al. 1993) and is a central player in post-transcriptional regulation of many transcripts, serving as a nuclear poly(A)-binding-protein regulating transcript stability (Schmid et al. 2015), poly(A) tail length, and poly(A) RNA export from the nucleus among other processes (reviewed in Moore 2005; Chen and Shyu 2014; and Stewart 2019). However, in metazoans Nab2 and ZC3H14 are dispensable for cellular viability, and the effects of either protein on poly(A) tail length and poly(A) RNA export from the nucleus are either less pronounced and likely exerted on fewer transcripts than in S. cerevisiae or are not detected (Farny et al. 2008; Kelly et al. 2010; Wigington et al. 2016; Bienkowski et al. 2017; Rha et al. 2017b; Morris and Corbett 2018). Consistently, Nab2/ZC3H14 do not associate with all polyadenylated RNAs tested to date in metazoans (Wigington et al. 2016; Bienkowski et al. 2017; Morris and Corbett 2018), but the possibility has remained that these few identified non-Nab2/ZC3H14-associated transcripts are outliers and that metazoan Nab2/ZC3H14 associates with a large majority of polyadenylated RNAs similarly to S. cerevisiae Nab2 (Tuck and Tollervey 2013), likely in part by binding poly(A) tails. Indeed, the identities of Nab2- or ZC3H14-associated RNAs in metazoans had never previously been addressed with a comprehensive, high-throughput method.
Our results identify a specific set of neuronal Nab2-associated mRNAs and noncoding RNAs in Drosophila. Of the 5760 total transcripts in the RIP-Seq testable set, only about 2.5% were found to associate with Nab2 in Drosophila neurons (Figure 5), a much smaller percentage of the transcriptome than associates with Nab2 in S. cerevisiae (Kim Guisbert et al. 2005; Batisse et al. 2009; Tuck and Tollervey 2013). Importantly, this likely represents an undercount of all Nab2-associated transcripts in neurons in vivo for two reasons: first, some RNAs associated with Nab2 in prior studies (Bienkowski et al. 2017) are not present in this Nab2-associated transcript set, likely due to technical limitations impacting sequencing read depth (see Materials and Methods), and second, our recent studies suggest that Nab2 can bind introns in pre-mRNAs (Jalloh et al. 2021), which may be more difficult to detect in a bulk RIP-Seq approach due to low abundance of pre-mRNAs relative to mature mRNAs. Higher sensitivity (e.g., CLIP-Seq) or more targeted approaches (e.g., sequencing IPs from pre-mRNA enriched samples) could thus reveal a broader set of Nab2-associated transcripts in Drosophila than defined here. Nonetheless, in the present study, the majority of cellular RNAs in the RIP-Seq testable set and a majority of tested polyadenosine-rich sequence motifs were not found to be associated with Nab2 (Figures 5 and 7), strongly supporting a model in which Nab2 interacts with a specific subset of RNAs in Drosophila neurons. Perhaps for this more select group of transcripts, Nab2 still plays a key role in transcript stability, poly(A) tail length control, transcription termination, and poly(A) RNA export from the nucleus, such that defects will only be observed in targeted examinations of single transcripts and not in bulk assays. This model of Nab2 specificity in Drosophila aligns well with the knowledge that Nab2/ZC3H14 is essential for cellular viability in S. cerevisiae but not in Drosophila, mice, or, seemingly, humans (Anderson et al. 1993; Pak et al. 2011; Bienkowski et al. 2017; Rha et al. 2017b; Al-Nabhani et al. 2018; Wheeler et al. 2019). This diminished global requirement for Nab2/ZC3H14 in metazoans may be due, at least in part, to functional overlap with PABPN1, an evolutionarily distinct nuclear polyadenosine RBP that is absent in S. cerevisiae (Mangus et al. 2003) but controls poly(A) tail length and is essential in Drosophila (Benoit et al. 2005), mice (Vest et al. 2017), and human cells (Hart et al. 2015).
The model of Nab2 specificity in Drosophila does not conflict with its affinity for polyadenosine, which could theoretically allow Nab2 to bind all transcripts with a poly(A) tail. Even in S. cerevisiae, the broad binding profile of Nab2 (Batisse et al. 2009) and central role in poly(A) tail length control (Kelly et al. 2010), poly(A) RNA export from the nucleus (Green et al. 2002), and protection of poly(A) RNA from degradation (Schmid et al. 2015) does not translate to binding the poly(A) tails of all transcripts (Kim Guisbert et al. 2005; Tuck and Tollervey 2013). More broadly, linear sequence motifs alone are insufficient to explain RBP specificity; RBPs do not generally occupy all of their available binding motifs throughout the transcriptome (Li et al. 2010; Taliaferro et al. 2016). Moreover, nonparalog RBPs with substantially overlapping or identical linear target motifs still bind distinct RNA target sets, demonstrating that linear motifs are only one of a set of RNA features that direct RBP–RNA associations (Dominguez et al. 2018). Based on the present study, these general features of RBPs hold for Nab2 as well. MEME and FIMO motif analyses reveal a long A-rich motif and the canonical Nab2 binding motifs A12 and A11G are enriched in but not exclusive to Nab2-associated RNAs (Figure 6). Moreover, our previous proteomic analysis detected six pairs of protein homologs whose levels change in both Nab2 mutant fly heads and Zc3h14 mutant mouse hippocampi; significantly, a 29 bp A-rich motif is presented in all 12 of the corresponding mRNAs (Rha et al. 2017b; Corgiat et al. 2021). In sum, these data are consistent with a model in which Drosophila Nab2 exhibits A-rich binding specificity in vivo and may bind some but not all mRNA poly(A) tails despite its affinity for polyadenosine RNA in vitro (Pak et al. 2011).
Taken together, these data align with the model that in metazoans Nab2/ZC3H14 is more specific in its transcript associations than in S. cerevisiae. With this model in mind and the Nab2-associated transcripts identified in this study in hand, future research will be enabled to explore how Nab2 functions on these transcripts in Drosophila, and why this function is so crucial for adult viability, neuronal morphology, locomotion, and learning and memory. Given that a polyadenosine-rich motif along with A12 and A11G motifs are correlated with but are not sufficient for Nab2–RNA association, future research must also focus on what additional features of transcripts or their associated proteins promote or prevent Nab2 association.
Conclusion
In sum, the data we present here identify functional interactions between Nab2 and Atx2 in Drosophila brain morphology, adult viability, and circadian behavior and define a set of RNA transcripts associated with each protein in brain neurons. Crucially, these RNA sets overlap—some associated transcripts are shared between Nab2 and Atx2 and some are specific to each RBP. Moreover, testing a select subset of these associated RNAs in a candidate genetic screen reveals enrichment for factors that interact functionally with Nab2 in the developing eye. Identifying these RBP-associated transcripts provides potential mechanistic links between the roles in neuronal development and function their encoded proteins perform, Nab2, and Atx2. This foundation will be especially important for Nab2, as the exact molecular function of metazoan Nab2/ZC3H14 on the vast majority of its associated RNAs in any cell type remains largely unknown. The identity of many Drosophila Nab2-associated transcripts, now revealed, will be required to define Nab2/ZC3H14 function in metazoans and enable our understanding of why loss of this largely nuclear polyadenosine RBP results in neurological or neurodevelopmental deficits in flies and mice and in intellectual disability in humans.
Data availability
The authors affirm that all data necessary for confirming the conclusions of the article are present within the article and associated figures, tables, supplementary materials, and database accessions. Supplementary File S1 contains Supplementary Materials and Methods, including those focused on bulk Drosophila head isolation, immunoblotting, DESeq2-based count normalization, and Gene Ontology analyses. Supplementary File S2 contains detailed legends for Supplementary Tables S1–S4. Supplementary File S3 contains all custom code (i.e., both R and PRISM scripts) written to generate, analyze, or visualize data in this article and associated figures, tables, and supplementary materials. Sequencing data, including raw reads, processed counts, and statistical analyses for each individual RIP-Seq sample, are available at the Gene Expression Omnibus (GEO) under accession: GSE165677. Drosophila stocks are available upon request. Supplementary materials, including files, figures, and tables, are available at figshare: https://doi.org/10.25386/genetics.16574828. last accessed 21 October 2021.
Acknowledgments
The authors thank current and past members of the Moberg and Corbett lab groups, especially Drs. Ayan Banerjee, Rick Bienkowski, Daniel Barron, Binta Jalloh, Stephanie Jones, Annie McPherson-Davie, Milo Fasken, and Sara Leung for support, instruction, and discussion. We also thank Drs. Bing Yao, Jingjing Yang, Michael Christopher, and Carlos Moreno for initial bioinformatics advice, and the Georgia Genomics and Bioinformatics Core (GGBC) at the University of Georgia, especially Tyler James Simmonds and Dr. Magdy S. Alabady, for library preparation, sequencing, assistance in experimental design, and technical support. We thank Drs. Nancy Bonini and Michael Parisi for the gift of a Atx2X1 stock; Dr. Ravi Allada, Khadijah Hamid, and Dr. Satya Surabhi for the gift of a UAS>Atx2-3xFLAG stock; Dr. Chunghun Lim for the gift of rabbit α-Atx2; Dr. Corey S. Goodman for the contribution of 1D4 Anti-Fas2 to the DSHB; and Drs. Gary Bassell, Roger Deal, Steven Warren, James Q. Zheng, and their respective labs for assistance and use of their equipment; Laura Fox-Goharioon for confocal microscope training; Dr. Michael I. Love for extensive public online instruction in the methodology and use of DESeq2; Dr. Mauricio Rodriguez-Lanetty for a public TRIzol-column hybrid RNA extraction protocol; and Eileen Chow for public video instruction in bulk Drosophila head isolation. The authors also thank with broad enthusiasm the authors, contributors, and ongoing maintainers of the incredible public resources supporting this work and without which it would not have been possible, including Flybase (NIH U41HG000739, UK MRC MR/N030117/1), the Galaxy Project (NIH 2U41HG006620), the R Project, the Developmental Studies Hybridoma Bank (University of Iowa, NIH), and the Bloomington Drosophila Stock Center (NIH P40OD018537).
Funding
This research was funded by grants from the National Institutes of Health to J.C.R. (F31 HD088043) and A.H.C. and K.H.M. (R01 MH107305).
Conflicts of interest
The authors declare no conflicts of interest.
Literature cited
R Core Team.
R Studio Team.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}