





Abstract
Cryptococcus neoformans, the most common cause of fungal meningitis, is a basidiomycete haploid budding yeast with a complete sexual cycle. Genome modification by homologous recombination is feasible using biolistic transformation and long homology arms, but the method is arduous and unreliable. Recently, multiple groups have reported the use of CRISPR-Cas9 as an alternative to biolistics, but long homology arms are still necessary, limiting the utility of this method. Since the S. pyogenes Cas9 derivatives used in prior studies were not optimized for expression in C. neoformans, we designed, synthesized, and tested a fully C. neoformans-optimized (Cno) Cas9. We found that a Cas9 harboring only common C. neoformans codons and a consensus C. neoformans intron together with a TEF1 promoter and terminator and a nuclear localization signal (Cno CAS9 or “CnoCAS9”) reliably enabled genome editing in the widely used KN99α C. neoformans strain. Furthermore, editing was accomplished using donors harboring short (50 bp) homology arms attached to marker DNAs produced with synthetic oligonucleotides and PCR amplification. We also demonstrated that prior stable integration of CnoCAS9 further enhances both transformation and homologous recombination efficiency; importantly, this manipulation does not impact virulence in animals. We also implemented a universal tagging module harboring a codon-optimized fluorescent protein (mNeonGreen) and a tandem Calmodulin Binding Peptide-2X FLAG Tag that allows for both localization and purification studies of proteins for which the corresponding genes are modified by short homology-directed recombination. These tools enable short-homology genome engineering in C. neoformans.
Introduction
The opportunistic pathogenic yeast Cryptococcus neoformans is the most common cause of fungal meningitis and is responsible for ∼15% of deaths in AIDS patients, causing ∼200,000 deaths annually (Rajasingham et al. 2017). Cryptococcus neoformans is a basidiomycete yeast and thus highly diverged from the ascomycete model yeasts Saccharomyces cerevisiae and Saccharomyces pombe (Janbon et al. 2014). Originally classified as a single species with four serotypes (A–D), these serotype designations continue to evolve (Hagen et al. 2017), with the most common serotype studied (serotype A) now called C. neoformans with seven pathogenic species recognized. As a model organism, it has many advantages including ease of cultivation, stable haploid cells, and a complete sexual cycle (Chun and Madhani 2010). Excellent animal models of infection have been developed, enabling studies of host-pathogen interactions (Cox et al. 2000). A congenic strain pair, KN99α and KN99a, has been developed and is widely used in the field (Nielsen et al. 2003).
Facile genetic manipulation is a cornerstone of the study of microbial pathogens, allowing the use of reverse genetics to dissect gene function, discover novel virulence determinants, and identify novel drug targets. Developed in the 1990s, biolistic transformation can achieve efficient editing in C. neoformans (Davidson et al. 2002). Indeed, our laboratory has used this method to construct a genome-scale gene knockout collection which we have made available through the Fungal Genetic Stock Center (www.fgsc.net). However, drawbacks of the biolistic method include a requirement for long (1 kb) homology arms, an expensive biolistic instrument, costly gold nanoparticles, and other disposables. Moreover, biolistic transformation is finicky and not always reliable. Thus, there is an unmet need for a more facile method.
Several CRISPR-Cas9 systems have been developed for use in C. neoformans. In one system, biolistic delivery was used to introduce a self-processing sgRNA-encoding construct and a targeting construct harboring 1 kb homology arms into a strain already carrying CAS9 at the SH1 locus (Arras et al. 2016). Another system used electroporation of a “suicide cassette” in which recombinational events excise CRISPR-Cas9 components from a cassette containing both a Cas9/sgRNA segment and a homology flanked-selectable marker (Wang et al. 2016). Both methods achieved high (70–90%) efficiencies of editing when targeting the ADE2 locus. In the latter method, CAS9 and sgRNA cassettes were lost in approximately half the resulting transformants, permitting later reuse of their method (Wang et al. 2016). A more recent method dubbed TRACE (Transient CRISPR-Cas9 Coupled with Elecroporation) relies on transient expression of CAS9 and sgRNAs from separate linear DNA molecules introduced by electroporation together with a homology donor DNA (Fan and Lin 2018). TRACE achieved CAS9 and sgRNA cassette dose-dependent editing efficiencies between 40 to 90% when targeting the ADE2 locus. Most recently, two additional transient methods using electroporation were described, one using a multi-guide approach, and a second using CRISPR-Cas9 RNPs to achieve high editing efficiency (Wang 2018).
Despite their utility, prior implementations of CRISPR-Cas9 in C. neoformans share several limitations. First, they require at least 500 bp arms for homology-directed recombination, which require potentially difficult construction by cloning or fusion PCR. Second, they were not optimized for Cas9 expression in C. neoformans. For example, the TRACE system uses a Cas9 derivative that was codon optimized for C. elegans and contains two C. elegans introns. Such introns would not be optimal for splicing in C. neoformans (Burke et al. 2018), although introns are necessary for gene expression in C. neoformans (Goebels et al. 2013). We reasoned that additional efficiency gains from codon optimization and use of an optimal intron might allow for shorter homology or improve CRISPR-Cas9 editing efficiency in this pathogen. We report here that a C. neoformans-optimized (Cno) Cas9 enables short homology-directed genome engineering. We present a modification to the TRACE approach which allows genetic manipulation to be mediated by homology directed repair (HDR) involving short (50 bp) regions of homology with only transient expression of Cas9 and sgRNA components. Furthermore, we demonstrate that increased efficiency can be achieved using a strain in which Cas9 is constitutively expressed by prior integration into the genome and the sgRNA is expressed transiently after electroporation of a PCR product. Finally, we describe use of a sequence that encodes codon-optimized mNeonGreen with a tandem purification tag for C-terminal tagging of genes. These tools enable short-homology genome engineering for the first time in C. neoformans.
Materials and methods
Strains and media
All strains were maintained in 20% glycerol stocks stored at −70°C. Cryptococcus neoformans and Saccharomyces cerevisiae strains were recovered from frozen stocks on YPD media (2% Bacto Peptone, 2% dextrose, 1% yeast extract) with agar for 2–3 days at 30°C or directly in liquid YPD media overnight at 30°C in a roller drum incubator at 60 RPM. Precultures for yeast transformation and C. neoformans electroporation, as well as outgrowths to allow for recovery following electroporation were in liquid YPD media. Following electroporation, transformants were selected on YPD plates supplemented with either 125 μg/ml nourseothricin or 300 μg/ml hygromycin as appropriate. Screening for Ura- transformants was performed on solid YNB media (0.67% Difco yeast nitrogen base without amino acids, 2% dextrose, 2% agar for solid media) supplemented with 1 mg/ml 5-fluoroorotic acid (5-FOA) and necessary amino acids. For plasmid cloning experiments using gap repair in S. cerevisiae, transformants were selected on YNB media lacking uracil.
A list of strains, primers, and plasmids used in this study is provided in Supplementary Table S1.
Plasmid construction
CAS9 was optimized based on codon frequency in C. neoformans var. grubii strain H99. Specifically, we generated a codon table for each amino acid using all annotated H99 coding sequences. We identified the most common codon for each amino acid (Supplementary Table S2). In addition, we designed the codon optimized CAS9 gene to contain an intron from CNAG_05429. This intron was modified to have consensus 5′ and 3′ splice sites. This entire Cno CAS9 construct was ordered from GenScript (Piscataway, NJ, USA) through their gene synthesis service and was received as an insert in pUC57 flanked by XbaI and BamHI restriction sites. The pUC57-CAS9 plasmid was digested with XbaI and BamHI to liberate the CnoCAS9. The TEF1 promoter and terminator were amplified by PCR from H99 genomic DNA using primers P01 and P02 for the promoter, and P03 and P04 for the terminator, containing homology to the pRS316 multicloning site and CnoCAS9. The TEF1 promoter, optimized CAS9 construct, and TEF1 terminator were cloned into pRS316 digested with XbaI and HindIII by gap repair in S. cerevisiae strain BY4741. Saccharomyces cerevisiae transformations were performed by the lithium acetate procedure as described elsewhere (Gietz and Schiestl 2007). pRS316 DNA was recovered from resulting transformants using a Zymoprep Yeast Plasmid Miniprep Kit (Zymo Research, Irvine, CA, USA) and electroporated into Escherichia coli strain DH5α to isolate single plasmid copies. Plasmid was recovered from candidate E. coli using a NucleoSpin Plasmid, Mini kit (Macherey-Nagel, Düren, Germany) and verified by Sanger sequencing (Genewiz, South Plainfield, NJ, USA) using custom oligos P05-P12. One resulting correct clone was designated pBHM2403.
The HYGB marker was inserted upstream of CAS9 in pBHM2403 to yield pBHM2408. Briefly, HYGB was amplified from pBHM2402 (S. Catania, unpublished data) using primers P13 and P14 containing homology to the NotI and SacII sites of pRS316/pBHM2403. The HYGB marker on pBHM2402 contains the same ACT1 and TRP1 promoter and terminator sequences as the NAT1 marker. This PCR product was cloned into pBHM2403 digested with NotI and SacII by Gibson assembly using the NEB Hifi DNA Assembly Kit (New England Biolabs, Ipswich, MA, USA).
To generate a sgRNA expression cassette, the Cryptococcus native U6 promoter was amplified from JEC21 genomic DNA (Wang et al. 2016) using P15 and P16, containing homology to the sgRNA scaffold. The sgRNA scaffold followed by 6T terminator were amplified from pSDMA66 (Arras et al. 2016) using P17, containing homology to the U6 promoter, and P18. These PCR products were cloned by Gibson assembly into pRS316 digested with BamHI and HindIII. Candidate colonies were Sanger sequenced using universal primers M13_F and M13_R and an identified correct clone was designated pBHM2329.
The tagging construct template plasmid was generated by homologous recombination in yeast strain BY4741. Codon-optimized mNeonGreen, CBP-2X FLAG-UTR, and NAT1/HYGB segments were amplified from independent plasmids using primers P19 to P24 and transformed alongside pRS316 vector digested with NotI and ClaI. Yeast transformation was done using lithium acetate protocol followed by extraction of pRS316 plasmid from yeast cells as described (Hoffman and Winston 1987). Plasmid was isolated by transformation of DH5α by electroporation. Individual colonies were used for isolating plasmid and clones were verified by enzymatic digestion and gel electrophoresis followed by Sanger sequencing. Correct plasmids were designated as pBHM2404 (NAT1) and pBHM2406 (HYGB).
Electroporation
Electroporations were performed as described by Lin and colleagues with the following modifications (Fan and Lin 2018). Briefly, cultures were inoculated from overnight cultures into 100 ml YPD at an OD600 of 0.2 and grown for 4–5 h until an OD600 of 0.65–0.8 was reached. Cells were collected by centrifugation and washed twice in ice cold water, before being resuspended in electroporation buffer (10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 270 mM sucrose) (Fan and Lin 2018) and incubated at 4°C with 1 mM DTT for one hour. Cells were collected and resuspended in 250 µl of fresh ice-cold electroporation buffer for an approximate resulting cell-buffer slurry volume of 450–500 µl. Fifty microliters of cells were mixed with PCR products and transferred to a precooled 2 mm gap electroporation cuvette (Bio-Rad Laboratories, Hercules, CA, USA). A BTX Gemini X2 instrument was used for electroporation with the following settings: 500 V, 400 Ω, 250 µF. Cells were resuspended following electroporation in 1 ml YPAD and incubated at 30°C for 1 h before plating on selective media.
Primers, PCR products for deletion experiments
Primers were ordered from IDT using their 25 nmole DNA Oligo or 100 nmole DNA Oligo service as appropriate. Deletion cassettes for URA5 and ADE2 were amplified from pCH233 using primer pairs P25+P26, and P27+P28, respectively. Primer pairs contained approximately 50 bp of homology to the respective gene of interest. PCR was performed with ExTaq (Takara Bio Inc., Kusatsu, Shiga, Japan) per manufacturer’s instructions, supplemented with 2% DMSO. For transient Cas9 experiments, the CAS9 cassette was amplified from pBHM2403 using ExTaq per manufacturer’s instructions and primers M13_F and M13_R.
Cassettes expressing sgRNAs were amplified in two PCR steps. Briefly, a set of primer pairs containing the 20 bp target sequence was designed to amplify the U6 promoter and sgRNA scaffold with 6T terminator, respectively, from pBHM2329. U6 and scaffold products were then mixed in equal volumes and used as the template for fusion PCR using a cocktail of Pfu and Taq polymerases as previously described (Chun and Madhani 2010). For example, to produce the ADE2-1 sgRNA, the U6 promoter was amplified from pBHM2329 using M13_F andP30. The sgRNA scaffold and 6T were then amplified from pBHM2329 using P29 and M13_R. The two products were then joined by fusion PCR using primers P33 and P34. The URA5 sgRNA was produced similarly using primers P31 and P32.
The sizes of PCR products were verified by gel electrophoresis. All PCR products were purified using a Macherey-Nagel Gel and PCR clean-up kit following their PCR cleanup procedure and eluted in a small quantity of sterile ddH2O. Each PCR aliquot of 100 µl was cleaned in an individual column and eluted in 15–20 µl sterile ddH2O to ensure high DNA concentration.
Primers, PCR products for tagging experiments
Plasmid pBHM2404 (NAT1) was used as template for donor PCRs using primers with ∼50 bp homology to the genome sequence (primers P51 through P60). Each PCR product was verified by gel electrophoresis and purified using Macherey-Nagel Gel and PCR clean-up kit as described above. PCR products specifying sgRNA cassettes targeting downstream regions of each gene were also produced as described above, using primers P61 through P70.
Strain construction
To delete URA5 or ADE2, KN99α (CM026) was electroporated with 700 ng of URA5 or ADE2 sgRNA, 1 µg of CAS9 cassette, and 2 µg of the URA5 or ADE2 deletion cassette, as appropriate. CM2049 was constructed by electroporating KN99α (CM026) with 700 ng of SH2 sgRNA and 2 µg HYGB-CnoCAS9 amplified from pBHM2408 using primers M13_F and M13_R. HYGB-CAS9 was amplified using Q5 High-Fidelity DNA Polymerase (New England Biolabs) per manufacturer’s protocols. SH2 sgRNA was produced as described earlier using primers P47 and P48. Insertion of HYGB-CAS9 at the SH2 locus was verified by colony PCR using primers P49 and P50.
For tagging each of the CNAGs 04149, 00938, 05700, 05940, and 06400, CM2049 was electroporated with 700 ng of CNAG specific sgRNA and 2 µg of mNeonGreen-CBP-2xFLAG donor PCR product with CNAG specific 50 bp homology arms. Transformants were selected on YPAD + NAT plates. For deletion of CNAG_01050, CM2049 was transformed with 700 ng of CNAG_01050 sgRNA cassette and 2 µg of deletion cassette containing 50 bp of homology to CNAG_01050.
To assess protein level from different Cas9 expressing cassettes, Cas9 cassettes were transformed into KN99α. The Fraser lab Cas9 construct (pSDMA65) was digested with PacI and integrated at the SH1 locus by biolistic transformation (JF-Cas9). The Cas9 coding sequence from the Wang lab was cloned under the control of the TEF1 promoter and terminator in pRS316 vector followed by digesting it with NotI/KpnI to ligate it in pSDMA58 alongside a Hygromycin marker for selection and 1 kb homology arms to SH1 and integrated at the SH1 locus by digestion with PacI and biolistic transformation (PW-Cas9). Primers P87 through P94 were used for this construct. Primers P83 through P86 were used for genotyping transformants. The Lin lab CAS9 gene, under control of the GPD1 promoter and terminator, alongside a HYGB marker was PCR amplified from pXL1-Cas9 using Q5 polymerase and M13 forward and reverse primers. The CnoCAS9 construct was amplified from pBHM2408 as described for construction of CM2049. These two constructs were integrated at the SH1 locus by transforming CM026 with 2 µg of either Lin lab CAS9, or HYGB-CnoCAS9 alongside 700 ng of SH1 sgRNA cassette. The SH1 sgRNA was amplified as described above using primers P97 and P98. Transformants were genotyped using primers P95 and P96.
Colony PCR
A detailed stepwise protocol with images for Colony PCR is available in Supplementary File S1.
Briefly, cells were patched from a transformant colony onto fresh selective media. Next, a small (1–2 µl) quantity of cells was smeared evenly against the bottom of a PCR tube using a wooden toothpick. The cells were microwaved for 2 min, and PCR master mix was immediately added to the lysed cells. Colony PCR bands were assessed by gel electrophoresis in a 1% agarose gel.
Primers P39 + P41, P40 + P42 were used for PCR genotyping of the ade2 locus. Primers P44 + P45, P43 + P46 were used for PCR genotyping of the ura5 locus. Primers P71 + P72 through P76 were used for genotyping of their respective CNAG C-terminal tag candidates.
Genomic DNA purification and Sanger sequencing
Genomic DNA was purified by CTAB extraction. Briefly, cells from a 5 ml overnight culture were collected by centrifugation. Cells were washed once in sterile distilled water, following which the supernatant was entirely discarded and the pellet was frozen in liquid nitrogen or at −80°C. Five milliliters CTAB buffer [100 mM Tris pH 7.4, 0.7M NaCl, 10 mM EDTA pH 8.0, 1% CTAB (Sigma-Aldrich, St. Louis, MO, USA)] with 1% β-mercaptoethanol was prewarmed to 65°C and then added to the frozen pellet. The pellet was then incubated for 2 h at 65°C with periodic agitation to lyse the cells. DNA was then isolated by one or two rounds of chloroform extraction as necessary followed by isopropanol precipitation. The pellet following precipitation was resuspended in 400 µl TE buffer and treated with 1 µl of 10 mg/ml RNAse A with incubation at 37°C for 1 h. RNAse treated samples were then treated with 5 µl 20 mg/ml Proteinase K with incubation at 55°C for 1 h. DNA was then isolated again by phenol: chloroform extraction followed by ethanol precipitation. Pellets were resuspended in 100 µl distilled water.
Upstream and downstream junctions for the ADE2 ORF were amplified using primers P35 and P37, P36 and P38, respectively. PCR products were sent to Genewiz for Sanger sequencing using primers P41 or P42.
Murine infection model
All work was performed under an approved institutional animal protocol. Cryptococcus neoformans strains KN99α (CM026) and CM2049 were grown overnight in YPAD medium, washed twice in sterile saline, counted by hemocytometer, and resuspended to a concentration of 1 × 106 cells/ml in sterile saline. Groups of ten 6- to 12-week-old C57BL/6J mice were anesthetized by intraperitoneal injection of 75 mg/kg ketamine and 0.5 mg/kg dexmedetomidine. Mice were suspended by their front incisors from a silk thread, and 50 µl of yeast suspension (5 × 104 total yeast) were slowly pipetted into the nares. Mice were kept suspended for 10 min post inoculation, after which they were lowered, and anesthesia was reversed by intraperitoneal injection of 1 mg/kg atipamezole. Mice were monitored daily and, upon loss of 20% body weight relative to pre-infection or other signs of severe disease were euthanized by CO2 inhalation followed by cervical dislocation. Lung and brain tissue were harvested, resuspended to 5 ml final volume in sterile PBS, homogenized, and plated onto YPAD agar to determine CFUs. Statistical differences in survival and endpoint CFUs were assessed by the Mantel-Cox and Mann-Whitney tests, respectively, performed in GraphPad Prism version 9.2.0.
Fluorescence microscopy
Sample preparation
Five milliliters of YNB media supplemented with 2% glucose was inoculated with each tagged strain followed by incubation at 30°C.Log stage cultures were used for imaging. Two hundred microliters of culture suspension was centrifuged at maximum speed and the pellet was washed once with 200 µl PBS and pelleted again at maximum speed. The cell pellet was then resuspended in PBS with 100 µg/ml calcofluor white and incubated for 10–15 min in the dark at room temperature. Cells were pelleted again and washed once with PBS and once with sterile water. Finally, the cell pellet was resuspended in 50 µl sterile water and 10 µl of the cell suspension was spotted on glass slide and covered with coverslip for imaging.
Microscopy and image processing
Images were collected using Nikon ECLIPSE Ti2 microscope using an 100X oil immersion objective. Images were processed for background subtraction and color correction using NIS elements and ImageJ (Schneider et al. 2012).
Protein extraction and immunoblotting
Protein extraction was done using previously described protocols (Catania et al. 2020). In brief, two milliliters of culture at OD600 = 1 was collected by centrifugation, and the cell pellet was frozen in liquid nitrogen. The cell pellet was then resuspended in 200 µl of 10% TCA and incubated on ice for 10 min. Cells were collected by centrifugation and washed once with 100% acetone and air-dried for 10 min. The pellet was then resuspended in 80 µl 1M Tris (pH 8.0) and 200 µl 2x Lysis buffer (Genescript LDS sample buffer 4X) followed by addition of glass beads. Sample was then boiled for 5 min at 100°C followed by bead-beating twice for 90 s. The cell lysate was centrifuged to remove residual cells. Twenty-five microliters of sample was subjected to SDS-PAGE (Genescript SurePAGE™, Bis-Tris gel) and transferred to a nitrocellulose membrane. Blotting was performed using anti-FLAG M2 antibody(1:3000, cat# F3165, Sigma), Cas9 antibody (1:500, cat# 61758, Active motif), Histone H3 (1:2000, cat# PA5-16183, Invitrogen) in 5% milk in TBS-T (10 mM Tris-HCl pH 7.6, 150 mM NaCl, 0.1% Tween 20) at 4°C overnight followed by three washes of 10 min each in TBS-T. The membrane was then incubated with anti-mouse or anti-rabbit antibody conjugated to HRP (Bio-Rad) (1:8000 in 5% milk+TBS-T) for 60 min at room temperature followed by three washes, 10 min each in TBS-T. The membrane was then incubated in SuperSignal West Pico reagent (Thermo Scientific) for 5 min. and visualized using an Azure c400 instrument (Azure Biosystems).
Results
Expression optimization of Cas9 for C. neoformans
We reasoned that improved Cas9 expression in C. neoformans might allow for short homology (50 bp) mediated HDR. Codon optimization can increase gene expression, and introns are required for efficient expression in C. neoformans (Goebels et al. 2013). Accordingly, we engineered the CAS9 coding sequence to use the most common codon for each amino acid (as determined by using all predicted open reading frames), and further introduced the single intron from C. neoformans gene CNAG_05249 (Figure 1A). We chose this intron because it is average in length for C. neoformans (Burke et al. 2018), and we further modified the sequence to have a consensus 5′ splice site (GTATGT) and 3′ splice site (CAG). This was placed into the codon optimized CAS9 at an arbitrarily chosen position (after amino acid 348). The optimized CAS9 ORF was placed under the control of the TEF1 promoter and terminator; this promoter also contains an intron in its 5′ untranslated region. Finally, we added a sequence encoding two SV40 nuclear localization signals to the C-terminus of the CAS9 coding sequence. We refer to this gene as Cno CAS9 or CnoCAS9. To validate our optimization steps, we compared Cas9 protein expression level from CnoCAS9 against other published constructs by immunoblotting. CnoCAS9 shows considerably greater Cas9 expression than all other CAS9 versions not optimized for C. neoformans expression, including constructs using the same TEF1 promoter (Supplementary Figure S1).
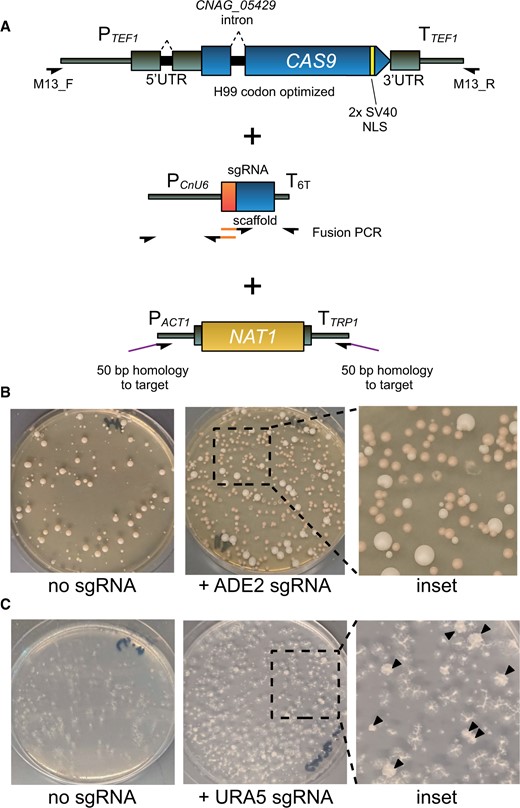
Optimized CRISPR enables short-homology editing in C. neoformans. (A) PCR scheme and schematic diagram for CAS9 cassette with relevant optimizations, sgRNA cassette, and short homology deletion cassette. Orange segments denote 20 bp guide sequences introduced by fusion PCR. (B) Transformation plates for deletion of ADE2 using transiently expressed optimized Cas9 and short homology deletion cassette. (C) Image of a URA5 deletion transformation plate replica plated onto 5-FOA media. Black arrows indicate colonies growing on 5-FOA.
Cryptococcus neoformans optimized Cas9 enables short homology-driven homologous recombination
To test our strategy for short homology-based editing, we targeted the ADE2 (CNAG_02294) locus, commonly used to assess the efficiency of gene editing techniques in fungi as the deletion strain accumulates a red pigment. We electroporated into KN99α linear DNAs corresponding to CnoCAS9, an ADE2 sgRNA expressing cassette (pU6-sgRNAADE2-1), and an ade2Δ::NAT1 deletion cassette with 50 bp of homology on either end to the ADE2 locus (amplified using ∼70 bp oligonucleotides). No red colonies were detected when pU6-sgRNAADE2-1 was omitted, whereas 69.4% of colonies from the electroporation that included pU6-sgRNAADE2-1 showed red pigmentation (Table 1, Figure 1B).
Colony counts of Ade- and Ura- transformants
| Experiment | Cas9 state | # aux − | # aux + | % aux − |
|---|---|---|---|---|
| ade2Δ | Transient | 174 | 76 | 69.6 |
| 158 | 69 | 69.6 | ||
| 129 | 58 | 69.0 | ||
| ura5Δ | Transient | 11 | 131 | 7.7 |
| 11 | 101 | 9.8 | ||
| 24 | 108 | 18.2 | ||
| ade2Δ | Integrated | 1730a | 4 | 99.8 |
| 1950a | 8 | 99.6 | ||
| 1520a | 7 | 99.5 | ||
| ura5Δ | Integrated | 82 | 53 | 60.7 |
| 106 | 17 | 86.2 | ||
| 69 | 22 | 75.8 |
| Experiment | Cas9 state | # aux − | # aux + | % aux − |
|---|---|---|---|---|
| ade2Δ | Transient | 174 | 76 | 69.6 |
| 158 | 69 | 69.6 | ||
| 129 | 58 | 69.0 | ||
| ura5Δ | Transient | 11 | 131 | 7.7 |
| 11 | 101 | 9.8 | ||
| 24 | 108 | 18.2 | ||
| ade2Δ | Integrated | 1730a | 4 | 99.8 |
| 1950a | 8 | 99.6 | ||
| 1520a | 7 | 99.5 | ||
| ura5Δ | Integrated | 82 | 53 | 60.7 |
| 106 | 17 | 86.2 | ||
| 69 | 22 | 75.8 |
Values extrapolated from colony counts of a 1:100 dilution of transformed cells.
Colony counts of Ade- and Ura- transformants
| Experiment | Cas9 state | # aux − | # aux + | % aux − |
|---|---|---|---|---|
| ade2Δ | Transient | 174 | 76 | 69.6 |
| 158 | 69 | 69.6 | ||
| 129 | 58 | 69.0 | ||
| ura5Δ | Transient | 11 | 131 | 7.7 |
| 11 | 101 | 9.8 | ||
| 24 | 108 | 18.2 | ||
| ade2Δ | Integrated | 1730a | 4 | 99.8 |
| 1950a | 8 | 99.6 | ||
| 1520a | 7 | 99.5 | ||
| ura5Δ | Integrated | 82 | 53 | 60.7 |
| 106 | 17 | 86.2 | ||
| 69 | 22 | 75.8 |
| Experiment | Cas9 state | # aux − | # aux + | % aux − |
|---|---|---|---|---|
| ade2Δ | Transient | 174 | 76 | 69.6 |
| 158 | 69 | 69.6 | ||
| 129 | 58 | 69.0 | ||
| ura5Δ | Transient | 11 | 131 | 7.7 |
| 11 | 101 | 9.8 | ||
| 24 | 108 | 18.2 | ||
| ade2Δ | Integrated | 1730a | 4 | 99.8 |
| 1950a | 8 | 99.6 | ||
| 1520a | 7 | 99.5 | ||
| ura5Δ | Integrated | 82 | 53 | 60.7 |
| 106 | 17 | 86.2 | ||
| 69 | 22 | 75.8 |
Values extrapolated from colony counts of a 1:100 dilution of transformed cells.
We next targeted URA5 (CNAG_03196) using the same method. We transformed KN99α with linear DNAs corresponding to CnoCAS9, the sgRNA (pU6-sgRNAURA5-1), and the ura5Δ::NAT1 deletion cassette. Transformants were replicaplated onto media containing 5-fluoroorotic acid (5-FOA) to screen for Ura- mutants (Boeke et al. 1984). Although the efficiency of deletion was lower than for ADE2, we still readily obtained the desired transformants, with 11.9% of colonies growing when replica plated on 5-FOA (Table 1, Figure 1C). The lower efficiency may be due to differences in sgRNA efficiency, or accessibility of the URA5 locus to Cas9, but low editing efficiency was observed even when additional sgRNA targets were tested (data not shown).
Although these data demonstrate that our optimized Cas9 system can be used to disrupt genes of interest, it was unclear if the mutants arose by HDR through 50 bp homology. Previous attempts with short homology using TRACE only disrupted the target locus through insertion of the marker, presumably via nonhomologous end-joining (Fan and Lin 2018). We predicted three potential resolutions of a Cas9 induced double stranded break (DSB): recombination could occur at both ends to yield full deletion/replacement; nonhomologous end joining (NHEJ) could occur at both ends resulting in an insertion mutant; or recombination could occur at one end, but NHEJ could occur at the other end, resulting in a partial deletion (Figure 2A). To determine whether the Ura- and Ade-transformants were insertion or deletion mutants, we designed PCR primers to test whether the desired HDR event had occurred at each end. PCR products should only be amplified if insertion via NHEJ occurred. We used this approach to assess transformants from three replicates each of transformations targeting ADE2 and URA5: 16 red colonies from each ade2Δ transformation and all colonies from ura5Δ transformations that grew on 5-FOA plates. Half of the ade2 mutants underwent HDR at either the 5′ or 3′ end of the ade2 ORF, and 61% of the ura5 mutants underwent HDR at either end (Table 2; representative colony PCR results are shown in Figure 2B). Among these transformants, we identified multiple ade2 and ura5 mutants that produced no PCR products from either the 5′ or 3′ ends of the targeted ORF, consistent with a full ORF deletion (Figure 2B). We selected eight such ade2 mutants for further analysis by Sanger sequencing of the upstream and downstream regions of the putative ade2Δ::NAT1 ORF (amplified by PCR using primers annealing 450 bp upstream or downstream of the recombinational junctions and within the NAT1). This sequencing showed that both junctions from all eight mutants were consistent with precise HDR (Figure 2C). These data demonstrate that the CnoCAS9 system allows for precise deletion using 50 bp homology arms.
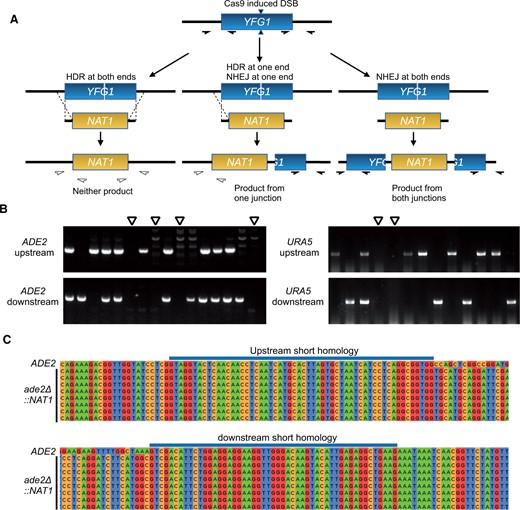
Genotyping of ade2 and ura5 transformants. (A) Possible outcomes of recombination outcomes and colony PCR genotyping scheme. Filled half-arrows indicate primer pairs that should amplify the target region. (B) Representative gels from colony PCR genotyping of transformants. Open arrowheads indicate candidates lacking both 5′ and 3′ junctions, consistent with the expected patterns for full ade2Δ and ura5Δ mutants. (C) Sanger sequencing of ade2Δ candidates from colony PCR genotyping.
Colony PCR results by experiment
| Experiment | Cas9 state | Total colonies tested | 5′ HDR only | 3′ HDR only | Both |
|---|---|---|---|---|---|
| ade2Δ | Transient | 48 | 7 | 6 | 11 |
| ade2Δ | Integrated | 48 | 14 | 5 | 6 |
| ade2Δ + W7 | Transient | 48 | 4 | 0 | 21 |
| ura5Δ | Transient | 26 | 1 | 5 | 10 |
| ura5Δ | Integrated | 31 | 4 | 3 | 5 |
| ura5Δ + W7 | Transient | 22 | 3 | 3 | 2 |
| Experiment | Cas9 state | Total colonies tested | 5′ HDR only | 3′ HDR only | Both |
|---|---|---|---|---|---|
| ade2Δ | Transient | 48 | 7 | 6 | 11 |
| ade2Δ | Integrated | 48 | 14 | 5 | 6 |
| ade2Δ + W7 | Transient | 48 | 4 | 0 | 21 |
| ura5Δ | Transient | 26 | 1 | 5 | 10 |
| ura5Δ | Integrated | 31 | 4 | 3 | 5 |
| ura5Δ + W7 | Transient | 22 | 3 | 3 | 2 |
Colony PCR results by experiment
| Experiment | Cas9 state | Total colonies tested | 5′ HDR only | 3′ HDR only | Both |
|---|---|---|---|---|---|
| ade2Δ | Transient | 48 | 7 | 6 | 11 |
| ade2Δ | Integrated | 48 | 14 | 5 | 6 |
| ade2Δ + W7 | Transient | 48 | 4 | 0 | 21 |
| ura5Δ | Transient | 26 | 1 | 5 | 10 |
| ura5Δ | Integrated | 31 | 4 | 3 | 5 |
| ura5Δ + W7 | Transient | 22 | 3 | 3 | 2 |
| Experiment | Cas9 state | Total colonies tested | 5′ HDR only | 3′ HDR only | Both |
|---|---|---|---|---|---|
| ade2Δ | Transient | 48 | 7 | 6 | 11 |
| ade2Δ | Integrated | 48 | 14 | 5 | 6 |
| ade2Δ + W7 | Transient | 48 | 4 | 0 | 21 |
| ura5Δ | Transient | 26 | 1 | 5 | 10 |
| ura5Δ | Integrated | 31 | 4 | 3 | 5 |
| ura5Δ + W7 | Transient | 22 | 3 | 3 | 2 |
Although 50 bp homology was sufficient to induce HDR, we frequently observed partial deletions with HDR at only one end. Arras and colleagues previously reported that supplementation of pre-culture media with W7 hydrochloride, an inhibitor of nonhomologous end-joining, increased the overall frequency of homologous recombination in the context of biolistic transformation (Arras and Fraser 2016). We reasoned that similar inhibition of NHEJ might result in increased HDR events. We tested this by adding 10 μg/ml W7 to both the preculture and outgrowth media for electroporation while targeting the ADE2 locus for deletion as previously described. Colony PCR showed that while including W7 did not change overall transformation efficiency, it did increase the co-occurrence of HDR in the same transformant (Supplementary Figure S2). Without W7, 45% (11/24) of colonies that underwent HDR showed a full deletion, compared to 84% (21/25) with W7 supplementation (n = 3, P = 0.015, Student’s t-test) (Table 2). We did not observe any change in HDR frequency for deletion of URA5. These data show that W7 supplementation may increase HDR, but that this effect may be sgRNA- and/or locus-dependent.
Stable integration of CnoCAS9 increases transformation and homologous recombination efficiency
We observed low transformation efficiency when we attempted to generate a URA5 deletion, despite testing multiple distinct sgRNA-encoding cassettes (data not shown). Fan and colleagues have reported that increasing the concentration of CAS9-encoding DNA or sgRNA-encoding construct improved both editing and transformation efficiency (Fan and Lin 2018) and Arras and colleagues previously found that their system required a genome-borne copy of the CAS9 gene to function effectively (Arras et al. 2016). Based on these published findings, we reasoned that a genome-borne copy of CnoCAS9 may permit greater efficiency in short-homology mediated manipulation at difficult loci, both due to an increase in CnoCAS9 expression from a stably integrated copy, as well as a reduction in the number of separate DNA molecules that a given cell would need to take up during transformation. Two loci have been reported to be “Safe Havens” for integration of transgenes in C. neoformans, Safe Haven 1 (SH1) and Safe Haven 2 (SH2), where gene insertion does not impact fungal virulence (Arras et al. 2015; Upadhya et al. 2017). We inserted a CnoCAS9 marked with HYGB at the SH2 locus using TRACE without any flanking homology (see Materials and Methods) and confirmed integration by colony PCR of this construct at the SH2 locus.
This resulting strain, CM2049 showed drastically increased editing efficiency (Figure 3). Transformation efficiency increased from approximately 300 colonies per ug DNA using transiently expressed Cas9 to more than 1 × 105 colonies per µg DNA with CM2049. Furthermore, when transforming CM2049 to delete ADE2, nearly all transformant colonies showed red pigmentation (Figure 3A). For URA5 deletion in CM2049, 74.2% of transformants grew on media containing 5-FOA (Table 1). Colony PCR of Ade- and Ura- transformants showed that several transformants had undergone HDR at both ends (Figure 3B). Across 3 replicates, 7.4% of all transformants from URA5 deletion experiments underwent homologous recombination at both junctions to delete URA5 using transiently expressed CnoCAS9, compared to 30.8% using genome-borne Cas9.
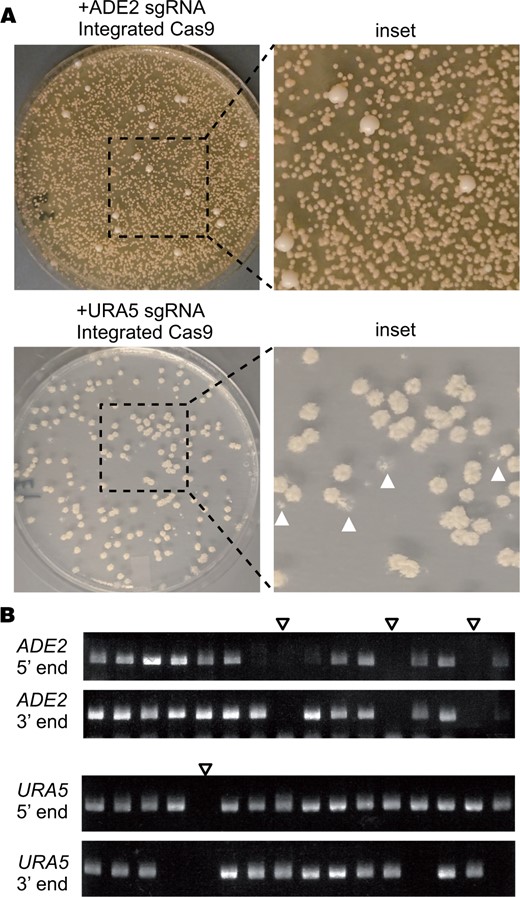
Deletion experiments using a genome-borne copy of optimized CAS9. (A) Transformation plates for deletion of ADE2 or URA5 in CM2049 using a short homology deletion cassette. The URA5 plate shows transformants replica plated onto 5-FOA media. White arrowheads indicate examples of colonies that failed to grow on 5-FOA. (B) Representative gels from colony PCR genotyping of ade2 and ura5 transformants in CM2049. Empty arrowheads indicate candidates lacking both 5′ and 3′ junctions, consistent with the expected patterns for full ade2Δ and ura5Δ mutants.
Integration of CnoCAS into the C. neoformans genome does not impact mammalian virulence
To test whether the strain harboring integrated CnoCAS9 impacted infection and virulence, we inoculated groups of 10 C57BL/6J mice intranasally (Cox et al. 2000) with the parental KN99α strain or the derivative expressing CnoCAS9 (CM2049). Infection was allowed to proceed until mice reached their predefined experimental endpoint (see Materials and Methods). Survival curves of both groups of mice were similar (Figure 4A) and there were no significant differences in lung or brain fungal burden at the time of sacrifice (Figure 4, B and C), indicating that integration of CnoCAS9 did not influence fungal virulence or dissemination to the central nervous system (Figure 4).
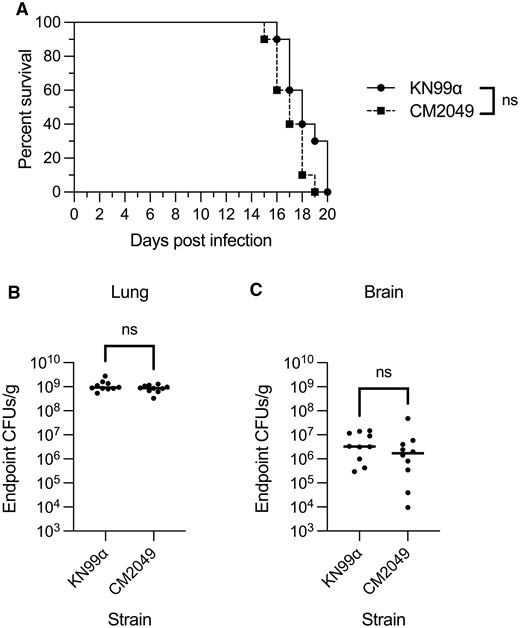
Evaluation of CM2049 infectivity in mice. Groups of 10 C57BL/6J mice were infected with 5 × 104 cells of either CM2049 or its parent strain KN99α. (A) Kaplan-Meier survival curve. ns, not significant, Mantel-Cox test. (B and C) CFUs per gram of organ tissue in the lungs (B) and brains (C) of mice at the time of sacrifice. ns, not significant, Mann-Whitney test.
Design and implementation of a localization and purification tag
Using the codon optimization approach described above, we generated and tested optimized versions of several fluorescent proteins, including GFP, mCherry, and mNeonGreen. We found that a Cryptococcus-optimized mNeonGreen performed particularly well. We attached to this sequence a calmodulin-binding protein tag followed by 2X FLAG purification tag we have used in prior work (Dumesic et al. 2013, 2015a, 2015b; Burke et al. 2018; Catania et al. 2020; Summers et al. 2020) to produce a combined localization and purification (LAP) tag. We also appended a cleavage-polyadenylation/transcription termination sequence from the CNAG_07988 gene and a NAT1 selectable marker (McDade and Cox 2001). Amplification of this construct with 50 bp of targeting homology enabled us to design donor DNAs to C-terminally tag genes of interest. In each case, we also encoded an sgRNA targeting a region just downstream of the coding sequence. We transformed CM2049 with a donor construct produced by PCR, and a guide-expressing construct (Figure 5). For one example, tagging the cryptococcal ortholog of the S. cerevisiae gene encoding the nucleoporin Nup107 (CNAG_04149) yielded a pattern consistent with the expected nuclear rim staining, and a protein product of the anticipated size of 132 kD (Figure 5).
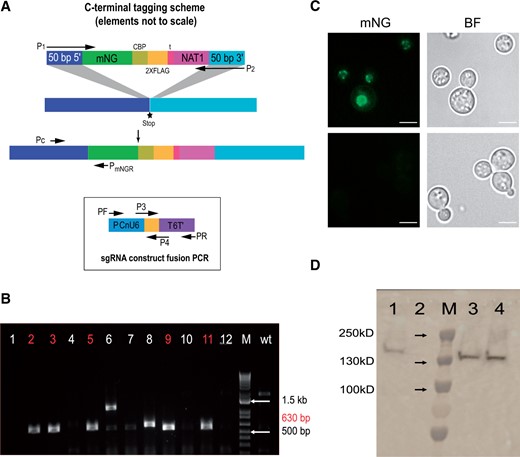
A LAP tag for C. neoformans (A) Scheme of the LAP-tagging strategy. (B) Colony PCR testing of 12 colonies and the parent strain to verify LAP tagging of NUP107. Positive colonies are highlighted in red. (C) Imaging of a positive clone from panel B, using a 100X oil immersion objective, FITC image and brightfield image at top and bottom panel is parental control image. (D) Immunoblot analysis. 1, positive control (CCC1-CBP-2X-FLAG, constructed by biolistic transformation); 2, parental strain; 3, NUP107-mNeonGreen-CBP-2XFLAG positive clone #1; 4, NUP107-mNeonGreen-CBP-2XFLAG positive clone #2.
To test our LAP tag in other cellular contexts, we tagged several additional genes selected based on information on their orthologs in S. cerevisiae or studies of localization in C. neoformans. These genes encode the mitochondrial co-chaperone Mrj1 (Horianopoulos et al. 2020), the plasma membrane ATPase Pma1 (Farnoud et al. 2014), the zinc finger transcription factor Cqs2 (Tian et al. 2018; Summers et al. 2020) and an ortholog of the S. cerevisiae endoplasmic reticulum chaperone Erj5 (Carla Fama et al. 2007). All of these yielded the expected localization patterns (Figure 6), suggesting that our tag will be a useful tool for studies of cryptococcal cell biology.
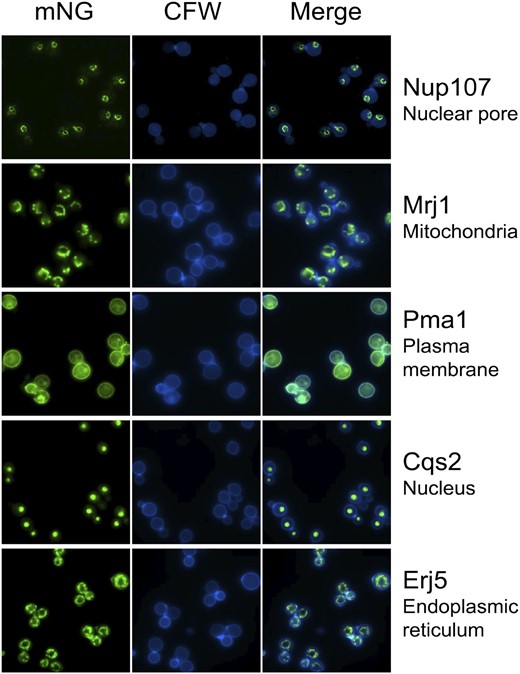
Proteins with distinct predicted localization patterns can be visualized with the C. neoformans LAP tag. Shown are representative images of strains expressing the indicated proteins tagged with mNeonGreen-CBP-2XFLAG; expected localization is included at the right. mNeonGreen fluorescence in the FITC channel is shown in the left column and calcofluor staining of cell wall chitin in the DAPI channel is shown in the center column.
Testing of portability and reproducibility
The tools described were developed in the Madhani laboratory. We sought to verify that our methods could be deployed in a different laboratory setting. We provided protocols and the CnoCAS9 expressing strain CM2049 to the Doering group, who successfully deleted CNAG_01050 and tagged NUP107 using these methods (Supplementary Figure S3). These data suggest that the tools described here are robust.
Discussion
Short homology-driven genome modification, first developed in S. cerevisiae (Baudin et al. 1993), enables rapid strain engineering, and was pivotal to the construction of whole-genome knockout and tag collections in S. cerevisiae (Giaever et al. 2002; Ghaemmaghami et al. 2003; Huh et al. 2003; Giaever et al. 2004). Cloning or fusion PCR to join long homology arms to a marker or construct of interest can be difficult and require extensive troubleshooting. Furthermore, a custom construct must be cloned for every desired manipulation, frustrating efforts to study multiple genes in parallel. Although CRISPR-Cas9 methods have been developed for use in C. neoformans, these methods have not addressed this major bottleneck.
In this study, we report a series of modifications to CAS9 to optimize Cas9 expression in C. neoformans. We show that the optimized CAS9 gene successfully allows use of short homology for genetic manipulation, results that have been replicated by a second group. Our optimized version represents an improvement over current CAS9 genes used in Cryptococcus research and, consistent with previous observations by Arras and colleagues for the human-optimized Cas9 (Arras et al. 2016), integration of this protein does not alter fungal virulence. Although our construct was optimized specifically for C. neoformans, we expect that it may be of broader utility, given the previously observed activity of Caenorhabditis elegans codon optimized Cas9 activity in other Cryptococcus species (Fan and Lin 2018).
Another approach to improving short homology-based editing in species that display high levels of NHEJ is the incorporation of single-stranded DNA strategies. Single-stranded DNA oligonucleotides (ssODN) were first shown to increase editing efficiency when using zinc-finger nucleases in human cell lines (Chen et al. 2011). With long or short ssODNs, editing in induced pluripotent stem cells, rats, and zebrafish has been achieved using 30, 60, and 40 bp homology arms respectively (Bai et al. 2015; Yoshimi et al. 2016; Okamoto et al. 2019). An efficient 100 bp homology arm knock-in strategy for Drosophila has been described that generated long ssDNA cassettes by enzymatic digestion of a PCR product with one 5’ phosphorylated strand (Kanca et al. 2019). Likewise, 120 bp single stranded homology arms on a hybrid donor can produce efficient editing in C. elegans (Dokshin et al. 2018). Although these approaches were not required for successful short homology editing in C. neoformans, they may allow further increases in efficiency. Initial efforts to transform KN99α with ssDNA by electroporation were not successful (data not shown), but further optimizations may open this promising avenue.
Data availability
Strains and plasmids are available upon request. Plasmids and plasmid sequences for pBHM2403, pBHM2404, pBHM2406, and pBHM2329 are available through Addgene. The authors affirm that all data necessary for confirming our findings are represented in the article and its tables and figures.
Supplementary material is available at GENETICS online.
Acknowledgments
The authors would like to thank Dr. Sandra Catania for her extensive advice and the members of the Madhani lab for many insightful and helpful suggestions. They also thank Dr. Stefan Oberlin for the kind gift of Cas9 antibody.
Funding
This work was supported by National Institutes of Health grants T32HL007185 (M.J.B.), F32AI152270 (M.J.B.), F31AI150194 (L.C.L.), T32GM007067 (E.A.G.), R01AI135012 (T.L.D), R01AI087794 and R01AI000272 (H.D.M.)
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
Author notes
Manning Y. Huang and Meenakshi B. Joshi are cofirst authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}