





Abstract
Continuously dividing cells coordinate their growth and division. How fast cells grow in mass determines how fast they will multiply. Yet, there are few, if any, examples of a metabolic pathway that actively drives a cell cycle event instead of just being required for it. Here, we show that translational upregulation of lipogenic enzymes in Saccharomyces cerevisiae increased the abundance of lipids and promoted nuclear elongation and division. Derepressing translation of acetyl-CoA carboxylase and fatty acid synthase also suppressed cell cycle-related phenotypes, including delayed nuclear division, associated with Sec14p phosphatidylinositol transfer protein deficiencies, and the irregular nuclear morphologies of mutants defective in phosphatidylinositol 4-OH kinase activities. Our results show that increased lipogenesis drives a critical cell cycle landmark and report a phosphoinositide signaling axis in control of nuclear division. The broad conservation of these lipid metabolic and signaling pathways raises the possibility these activities similarly govern nuclear division in other eukaryotes.
Introduction
The sequential and essential action of acetyl-CoA carboxylase (Acc1p in yeast) and fatty acid synthase (FAS) make fatty acids in all organisms. Acc1p carboxylates acetyl-coenzyme A (CoA) to make malonyl-CoA (Al-Feel et al. 1992), subsequently consumed by FAS as the building block for fatty acid synthesis. Yeast FAS is made of two different polypeptides, α- (Fas2p) and β-subunits (Fas1p), in a 2.6 MDa α6β6 dodecamer (Jenni et al. 2007; Lomakin et al. 2007; Leibundgut et al. 2008). The correct Fas1p and Fas2p stoichiometry is achieved by a mechanism where Fas1p controls FAS2 mRNA levels (Wenz 2001). Moreover, Fas1p initiates FAS assembly via a cotranslational interaction with nascent Fas2p (Shiber et al. 2018; Fischer et al. 2020). The multiple catalytic activities of FAS iteratively add two carbons at a time from a malonyl-CoA donor to an acetyl-CoA acceptor. The reaction terminates after seven or eight cycles, generating 16- or 18-carbon fatty acids, respectively (Jenni et al. 2007; Lomakin et al. 2007).
There is intense contemporary interest in lipogenesis during eukaryotic cell division, given the implications for cell proliferation (Storck et al. 2018). Higher levels of lipogenic enzymes are a near-universal marker of tumors (Kuhajda et al. 1994; Baenke et al. 2013; Beloribi-Djefaflia et al. 2016), and FAS is one of the few metabolic enzymes targeted in cancer clinical trials (NCT02595372 and NCT02980029). Based on correlative data, it is possible that de novo lipid synthesis impinges on events late in the cell cycle. First, the translational efficiency of the acetyl-CoA carboxylase and FAS mRNA transcripts peaks in mitosis (Blank et al. 2017a, 2017b). Second, the abundance of lipids similarly peaks late in the cell cycle (Blank et al. 2020). We also note that the midbodies in human cells, where cleavage occurs at the end of the cell cycle, have a distinct lipid composition (Atilla-Gokcumen et al. 2014). Third, de novo lipid synthesis is essential for mitosis in humans (Scaglia et al. 2014) and yeast (Schneiter et al. 1996; Al-Feel et al. 2003).
Nuclear membrane dynamics are pronounced during mitosis. For example, there is a dramatic proliferation in new nuclear membranes during mitosis. This proliferation occurs irrespective of whether the mitosis is open (i.e., the nuclear envelope disassembles in mitosis as in animal cells) or closed (i.e., nuclear envelope integrity is maintained throughout mitosis as in fungi). In fission and budding yeast, loss-of-function mutations in lipid synthesis perturb the nuclear envelope’s size and shape (Santos-Rosa et al. 2005; Walters et al. 2012, 2014; Witkin et al. 2012; Siniossoglou 2013; Kume et al. 2017; Zach and Převorovský 2018). The evidence that lipid synthesis is required for cell division notwithstanding, it remains unclear whether lipid biogenesis promotes specific cell cycle events and, if so, what those events might be.
Sec14p is the founding member of a major family of phosphatidylinositol (PtdIns) transfer proteins (PITPs). Sec14p and other PITPs stimulate the activities of PtdIns 4-OH kinases by rendering PtdIns a superior substrate for these enzymes—thereby potentiating PtdIns(4)P-dependent signaling (Wang et al. 2019). Sec14p executes an essential cellular activity in promoting membrane trafficking from yeast Golgi/endosomal compartments, but this cellular requirement is obviated when lipid metabolism is appropriately altered. For example, the inactivation of the CDP-choline salvage pathway for PtdCho synthesis relieves the essential Sec14p requirement (Cleves et al. 1991). In addition to membrane trafficking, Sec14p also controls cell cycle progression (Huang et al. 2018). Reductions in Sec14p activity of insufficient magnitude to compromise cell viability result in larger cells that progress through the cell cycle more slowly, with notable delays in progression through the G2/M phases (Huang et al. 2018). How Sec14p impinges on cell cycle progression and how it is integrated with lipid synthesis in the cell cycle remains poorly understood.
Here, we demonstrate that coordinate derepression of ACC1 and FAS1 translation results in elevated lipogenesis that promotes nuclear division in yeast. Furthermore, we report that loss-of-function mutations in the conserved Sec14p delay nuclear division and that derepressing translational control of ACC1 and FAS1 not only corrects the delayed nuclear division associated with reduced Sec14 activity but also suppresses other mitotic phenotypes associated with Sec14p insufficiencies. Likewise, we document that loss of phosphatidylinositol 4-OH kinase activity deranges nuclear morphology and that these phenotypes too are rescued by enhanced translation of ACC1 and FAS1 mRNAs. These results identify translational control as a critical input for tuning lipid metabolism to progression through the cell cycle. The data also indicate that lipid synthesis is not only required for mitosis, but that it actively promotes nuclear division, a key landmark of the eukaryotic cell cycle.
Materials and methods
A Reagent Table is in Supplementary material. Where known, the Research Resource Identifiers are shown in Supplementary Reagent Table.
Strains and media
All the strains used in this study are shown in Supplementary Reagent Table. Unless noted otherwise, the cells were cultivated in the standard, rich, undefined medium YPD [1% (w/v) yeast extract, 2% (w/v) peptone, and 2% (w/v) dextrose], at 30°C (Kaiser et al. 1994). To modify the FAS1-5′ leader, we first inserted a URA3 gene (amplified from a prototrophic strain X1280) upstream, at position ChrXI99751 with the PCR-mediated methodology of (Longtine et al. 1998), using primers XI99701-URA-F1 and XI99800-URA-R1 (see Supplementary Reagent Table). The PCR product was transformed into a yeast strain carrying the TAP epitope at the 5′ end of the FAS1 main ORF (FAS1-TAP, see Supplementary Reagent Table). The resulting strain, NM21, was genotyped by PCR for the presence of the insertion at the expected location, using primers (XI99514-F1 and XI99895-R1) that flank the insertion. Genomic DNA of NM21 was then used as a template in a PCR reaction with the forward primer XI99514-F1 and reverse primers FAS1-141-REV or FAS1-279-REV, to introduce point mutations at the start codon of the proximal uORF (T141A) or the distal uORF (G279A), respectively. The strains obtained were mutant for the proximal (NM19) or distal uORF (NM24). The introduced mutations were verified by PCR (with primers XI100101-F1 and XI100698-R1) followed by restriction digestion with enzymes PsiI (for the proximal uORF mutation) and RsaI (for the distal uORF mutation). The T141A mutation introduces a restriction site for PsiI at the proximal uORF. On the other hand, the G279A mutation eliminated the RsaI restriction site at the distal uORF. Next, to obtain a double uORF FAS1 mutant, the genomic DNA of NM24 was used as a template in a PCR reaction with forward primer XI99514-F1, and reverse primer FAS1-141-REV. The resulting product was transformed into FAS1-TAP cells, to generate strain NM31, which was verified by PCR, followed by restriction digestion PsiI. The introduced uORF mutations were also confirmed by sequencing the strains’ genomic DNA using the primers XI100101-F1 and XI100698-R1.
To derepress translation of both ACC1 and FAS1 in the same cells, we crossed strain NM31 with the ACC1-uORF mutant [carrying the TG340AA substitutions in the 5′-leader of ACC1; strain HB1, see Supplementary Reagent Table and Blank et al. (2017a)], followed by sporulation and tetrad dissection. The resulting strains (NM36 and NM35) have point mutations at the start codons of the uORFs of both ACC1 and FAS1 (uORFm-ACC1 and FAS1). To obtain wild-type (WT) strains with markers matching those of uORFm-ACC1, FAS1 cells, we crossed NM21 with HB13 [ChrXIV 662143::kanMX6, see Blank et al. (2017a)], followed by sporulation and tetrad dissection, yielding strains NM33 and NM34. Unless indicated otherwise, strains NM33 and NM36 were used as the WT, and uORFm-ACC1, FAS1, strains shown in figures.
To generate the swe1Δ,uORFm-ACC1, FAS1 strain, we first sporulated the homozygous diploid swe1Δ/swe1Δ strain constructed by the yeast deletion project (Giaever et al. 2002), followed by tetrad dissection, to obtain the haploid swe1Δ strain (NM95). We then crossed strains NM35 and NM95, followed by sporulation and tetrad dissection, yielding strain NM92.
To introduce the sec14-1 allele, strains NM34 and NM35 were crossed with CTY1-1A (Xie et al. 2001), sporulated, and the tetrads dissected to generate the marker-matched sec14-1 (NM39), and the sec14-1, uORFm-ACC1, FAS1 quadruple mutant (NM42), respectively. Similarly, we generated all other mutant combinations shown in Figures 6 and 7.
Statistical analysis, sample-size, and replicates
For sample-size estimation, no explicit power analysis was used. All the replicates in every experiment shown were biological ones, from independent cultures. A minimum of three biological replicates was analyzed in each case, as indicated in each corresponding figure’s legends. Where three replicates were the minimum number of samples analyzed, the robust bootstrap ANOVA was used to compare different populations via the t1waybt function, and the post hoc tests via the mcppb20 function, of the WRS2 R language package (Wilcox 2011; Mair and Wilcox 2016). We also used nonparametric statistical methods, as indicated in each case. The Kruskal–Wallis and post hoc Nemenyi tests were done with the posthoc.kruskal.nemenyi.test function of the PMCMR R language package. No data or outliers were excluded from any analysis.
Immunoblot analysis
For protein surveillance, protein extracts were made as described previously (Amberg et al. 2006), and run on 4–12% Tris–glycine SDS-PAGE gels. To detect TAP-tagged proteins with the PAP reagent, we used immunoblots from extracts of the indicated strains, as we described previously (Blank et al. 2017a, 2020). Loading was evaluated with an anti-Pgk1p antibody. Images were processed with the ImageJ software package. Briefly, the “Subtract background” tool was applied, followed by the “Measure” tool to obtain a mean intensity for each band. The area measured was kept constant for a sample series for each blot analyzed.
Transcript abundance using ddPCR
For RNA surveillance, RNA extracts were made as described previously (Blank et al. 2017a). Briefly, RNA was extracted using the hot acidic phenol method (Collart and Oliviero 2001). Cell pellets were resuspended in 0.4 ml of TES buffer (10 mM Tris, pH 7.5; 10 mM EDTA, and 0.5% SDS) with 0.1 ml of glass beads. Then, 0.4 ml of acid phenol, pH 4.5, was added, and the samples were heated at 65°C for 0.5 h with occasional vortexing for 10 s each time. The samples were then centrifuged for 5 m at 13,000 g. The aqueous layer was transferred to a new tube containing 1 ml of cold 100% ethanol with 40 μl of 3 M sodium acetate and incubated overnight at 4°C. The next day, the samples were centrifuged at 13,000 g for 20 min at 4°C, washed with 80% ethanol, and centrifuged again for 5 min at 13,000 g. The pellets were resuspended in 25 μl water. The amount of total RNA in each sample was measured with a spectrometer. For the quantification of transcript abundance, 0.75 ng of total RNA was used for each sample.
The ddPCR reaction mixture was prepared by following the manufacturer’s protocol, using the Taqman hydrolysis probes labeled with FAM (for FAS1) and VIC (for UBC6) reporter fluorophores. The mixture was kept on ice throughout the whole experiment. Once the reaction mixture was prepared, the samples were placed into a droplet generator (QX200™ AutoDG™ Droplet Digital™ PCR System), which uses specially developed reagent and microfluidics to partition each sample into 20,000 nl-sized droplets. Once the droplets were generated, the samples were transferred to a thermal cycler (C1000 Touch™ Thermal cycler) for PCR. Following the PCR, the plate containing the droplets was placed in a droplet reader (Bio-Rad, QX200 Droplet reader). The autosampler of the droplet reader picks up droplets from each of the wells of the PCR plate, and fluorescence is measured for individual droplets.
The abundance of the transcripts was obtained using the QuantaSoft™ Software. Transcript levels of FAS1 were normalized against the corresponding transcript levels of UBC6.
Centrifugal elutriation, cell size, and DNA content measurements
All methods have been described previously (Hoose et al. 2012; Soma et al. 2014). Briefly, after early G1 cells were collected, they were monitored at regular time intervals for cell size and budding. Samples were also assayed in downstream procedures, such as nuclear staining, as described in the relevant sections.
Metabolite profiling
The untargeted, primary metabolite, and complex lipid analyses were done at the NIH-funded West Coast Metabolomics Center at the University of California at Davis, according to their established mass spectrometry protocols, as we described previously (Blank et al. 2020; Maitra et al. 2020). The raw data for the complex lipid measurements are in Supplementary File S1. The raw data for the primary metabolite measurements are in Supplementary File S2. To identify significant differences in the comparisons among the different strains, we used the robust bootstrap ANOVA, as described above. The input values we used were first scaled-normalized for input intensities per sample. Detected species that could not be assigned to any compound were excluded from the analysis.
Fluorescence microscopy
Cells were fixed with 3.7% (v/v) formaldehyde at room temperature for 30 m, washed three times with 0.1 M potassium phosphate buffer, followed by a wash and resuspension in 1.2 M sorbitol. Next, the fixed cells were digested with 0.06 mg/ml zymolyase 20 T for 5 m at 37°C, and later washed and resuspended again in 1.2 M sorbitol. Twenty microliters of the fixed and digested cells were added on a poly-lysinated slide and incubated at room temperature in a moist chamber for 20 min. The liquid was removed, and the slide was dehydrated by ice-cold methanol, and then acetone, for 3 min and 10 s, respectively. The slide was air-dried and incubated in 0.1% BSA/PBS for 30 min at room temperature. The BSA/PBS solution was removed, and the α-Nsp1 antibody (see Supplementary Reagent Table) was added at a dilution of 1:100 dilution and incubated overnight at 4°C. Nsp1p is a nucleoporin (Hurt 1988) used in our experiments to decorate the nuclear envelope (Supplementary Figure S4). The next day, the slide was washed five times with 0.1% BSA/PBS, followed by the addition of secondary antibody (Alexa Fluor 488 AffiniPure Goat α-Mouse IgG, see Supplementary Reagent Table) at a dilution of 1:100 and incubated at room temperature for 1 h. The slide was then washed with 0.1% BSA/PBS and mounted with the Vectashield mounting media containing DAPI to visualize the nuclear DNA.
Cells were viewed with a Nikon Eclipse TS100 microscope, with a 100× objective, and the images were captured with a CoolSnap Dyno 4.5 Nikon camera. The exposure time for the DAPI and GFP filters (Alexa Fluor 488) was 50 and 500 ms, respectively. All images were captured in NIS Elements Advanced Research (version 4.10) software. The fluorescent images acquired with the GFP and DAPI filters were processed in ImageJ. The ratio of two axes (long axis and the short axis) of the nuclear mass were measured using the “line” tool followed by the “measure” tool in the ImageJ software package. When the nucleus is near-spherical, the ratio is 1. However, as the nucleus starts to expand, the ratio increases, signifying the elongation of the nuclear envelope (Supplementary Figure S4; Figure 3).
Results
Translational control elements in lipogenic enzymes
The translational efficiency of acetyl-CoA carboxylase and FAS is upregulated late in the cell cycle, and removing a uORF from the 5′-leader of the ACC1 transcript derepresses the translation of ACC1 (Blank et al. 2017a). The 5′-leader of FAS1 is 551 nt-long (Xu et al. 2009) and has two uORFs (Figure 1A). Ribosome profiling experiments report that both FAS1 uORFs are likely translated in Saccharomyces cerevisiae (Ingolia et al. 2009). The longer and more distal uORF initiates from a non-AUG start codon. A uORF at the same position, albeit of varying length and sequence, is present in most Saccharomyces genomes, except in S. mikatae, where the AAG start codon is mutated to AAA (Supplementary Figure S1, top panel). The proximal uORF is short, encoding a six amino acid peptide, and it is well-conserved in the Saccharomyces genus (Supplementary Figure S1, bottom panel). There was a Tyr → Phe change in S. kudriavzevii, and a Phe → Leu change in S. bayanus (Supplementary Figure S1, bottom panel).
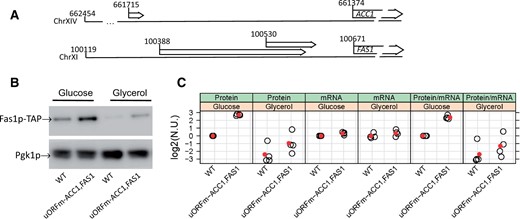
uORFs in ACC1 and FAS1. (A) The chromosomal coordinates of the 5′-leader sequences of the ACC1 and FAS1 transcripts. The 5′-ends are from Xu et al. (2009). (B) Steady‐state Fas1p protein levels were measured in rich undefined media, differing in the carbon source (2% glucose or 3% glycerol) from the indicated strains carrying C‐terminal TAP‐tagged alleles of FAS1 at their endogenous chromosomal locations. (C) The values in the strip charts depict the relative abundance of FAS1 mRNA and protein in uORFm-ACC1, FAS1 cells quantified from independent experiments, from the same strains and media shown in (B). The red symbols indicate the mean in each case. Transcript levels of FAS1 and UBC6 were quantified by Droplet Digital PCR (ddPCR), as described in Materials and Methods. To obtain the normalized units (n.u.) on the y‐axis, we first normalized for loading against the corresponding UBC6 and Pgk1p values from the same samples. We then expressed these values as ratios against the corresponding values of wild‐type FAS1‐TAP cells, in which the uORFs are in place, from experiments run and analyzed in parallel. All the immunoblots for this figure are in Supplementary File S3, while the mRNA abundance experiments we used to generate the strip plots are in Supplementary File S4.
To test the role of translational control in fatty acid synthesis, a mutant yeast strain was generated where cis-elements that could repress translation of ACC1 and FAS1 transcripts were inactivated. Acc1p and FAS act sequentially. Hence, simultaneous derepression of both ACC1 and FAS1 translation was expected to increase metabolic flux through de novo lipogenesis pathways. Because the FAS1 uORFs are likely translated in S. cerevisiae and are well conserved in other species, their start codons were mutated. The FAS1 uORF mutations were subsequently recombined into a yeast strain carrying the ACC1 ORF mutation we had described previously (Blank et al. 2017a). The strain carrying these three uORF mutations (uORFm-ACC1, FAS1) was also engineered so that the main FAS1 ORF was TAP-tagged for purposes of protein surveillance (see Materials and Methods). As expected, asynchronous cultures of the triple uORF mutant produced elevated steady-state levels of Fas1p (Figure 1B). The ratio of the steady-state levels of the Fas1p protein over the FAS1 mRNA was three to fourfold higher in the uORF mutant cells compared to WT cells (Figure 1C). These data were consistent with a translational derepression mechanism for increased Fas1p expression.
To monitor Fas1p levels throughout the cell cycle, small early G1 WT and uORFm-ACC1, FAS1 cells were isolated by centrifugal elutriation, and Fas1p-TAP levels were examined as these cells progressed synchronously through the cell cycle (Supplementary Figure S2). Fas1p-TAP levels still oscillated in uORFm-ACC1, FAS1 cells, albeit with dampened amplitude (Supplementary Figure S2). We note that Acc1p levels were similarly periodic, with dampened amplitude, in the cell cycle when ACC1 translation was derepressed in cells lacking the ACC1 uORF (Blank et al. 2017a).
Derepressing translation of ACC1 and FAS1 alters lipid abundances
We used two approaches to determine whether elevated levels of Acc1p and Fas1p in uORFm-ACC1, FAS1 cells led to increased activity of these enzymes in vivo. The first was a bioassay where the sensitivity of WT and triple mutant cells to cerulenin was compared. Cerulenin is a validated inhibitor of FAS that acts by binding and inhibiting the β-ketoacyl-acyl carrier protein synthase domain of FAS (Omura 1976; Price et al. 2001). Whereas proliferation of WT cells was significantly reduced and then completely blocked at 1.5 and 2.5 μg/ml cerulenin, respectively, the uORFm-ACC1, FAS1 cells were resistant to 2.5 μg/ml cerulenin (Supplementary Figure S3). The increased resistance of uORFm-ACC1, FAS1 cells to cerulenin argues that translational derepression of ACC1 and FAS1 leads to elevated activity of these lipogenic enzymes in vivo.
The second approach involved quantifying primary metabolites and complex lipids by GC-TOF MS, and CSH-QTOF MS/MS, respectively (see Materials and Methods). We examined cultures where glucose or glycerol served as sole carbon sources (Figure 2). Among the >300 metabolites that were confidently identified in cultures with glucose as the sole carbon source, the alterations in metabolite abundance were almost entirely accounted for by alterations in the abundance of lipid molecular species. Increased levels of various molecular species of triglycerides (TG), phosphatidylinositol (PtdIns; PI), phosphatidylcholine (PtdCho; PC), phosphatidylethanolamine (PtdEtn; PE), and phosphatidylserine (PtdSer; PS) were recorded. A few species exhibited a lower abundance in uORFm-ACC1, FAS1 cells and these represented the longer chain molecular species detected (PtdIns/PtdCho/PtdEtn 36:1; PtdCho 36:2; TG 50:1; TG 56:2). This pattern was reproduced in cultures where glycerol served as a carbon source, although the roster of molecular species altered in abundance in glycerol conditions was expanded somewhat. We conclude that coordinate derepression of ACC1 and FAS1 translation altered the lipidome predominantly by increasing the abundance of most lipid molecular species.
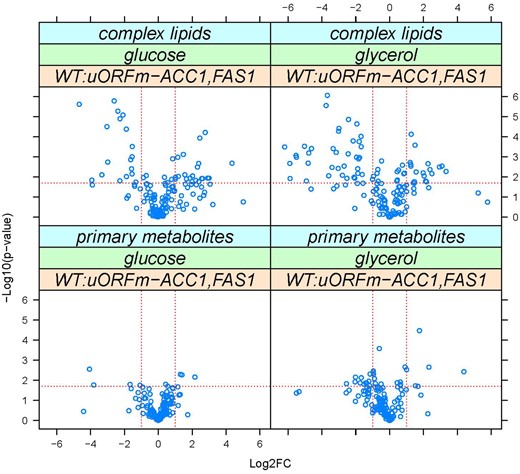
Derepressing the translational control of lipogenesis alters lipid abundances. Metabolites whose levels changed in WT vs uORFm-ACC1, FAS1 cells were identified from the magnitude of the difference (x-axis; Log2-fold change) and statistical significance (y-axis), indicated by the red lines. The analytical and statistical approaches are described in Materials and Methods. The values used to generate the graphs are in Supplementary File S6/Sheet2.
Increased lipogenesis promotes nuclear division
The uORFm-ACC1, FAS1 cells represent a unique, gain-of-function context for interrogating the effects of increased lipogenesis on nuclear division. It is well-established that lowering lipid synthesis perturbs the nuclear envelope’s shape in fungi (Santos-Rosa et al. 2005; Walters et al. 2012, 2014; Witkin et al. 2012; Siniossoglou 2013; Kume et al. 2017; Zach and Převorovský 2018). Those findings motivated examination of nuclear morphology in uORFm-ACC1, FAS1 cells progressing synchronously through the cell cycle. As in our past studies (Blank et al. 2017a, 2020; Huang et al. 2018), centrifugal elutriation was again employed to recover highly enriched populations of unarrested early G1 cells where the coupling between cell growth and division is maintained (Creanor and Mitchison 1979; Aramayo and Polymenis 2017). The timing of nuclear division was then compared for synchronous WT and uORFm-ACC1, FAS1 cultures.
To that end, the nuclear envelope was visualized by immunofluorescence using the Nsp1p component of the central core of the nuclear pore complex as a marker (see Materials and Methods and Supplementary Figure S4A). For each cell scored at one point in the cell cycle, the longest axis across the nucleus was related to the nonelongated, spherical nuclear space’s diameter and expressed as a ratio (Supplementary Figure S4A).
The cell size and percentage of budded cells from the same elutriated samples were also recorded. Since yeast bud emergence marks the initiation of cell division, this event serves as a convenient proxy for marking entry into the S phase (Pringle and Hartwell 1981). We therefore used this landmark to relate the initiation of cell division to cell size. A key parameter that derives from such an analysis is the critical size, defined as the size at which half the cells are budded (Soma et al. 2014). The data from four independent experiments are shown in Figure 3. The critical size of uORFm-ACC1, FAS1 cells was slightly smaller than the critical size of WT cells (Figure 3A; 36.7 ± 1.1 vs 40.6 ± 2.1 fl; n = 4; P = 0.03, based on the Mann–Whitney test). From the same elutriations, we calculate the specific rate of size increase by plotting the cell size data as a function of time. The rate at which uORFm-ACC1, FAS1 cells increased in size was also slightly lower than the rate for WT cells (0.28 ± 0.1 vs 0.32 ± 0.1/h, respectively; n = 4; P = 0.03, based on the Mann–Whitney test). Lastly, as we will show in a subsequent section using asynchronous cultures, the uORFm-ACC1, FAS1 cells are born at the same size as WT cells [21.5 ± 0.9 fl (n = 17) vs 21.3 ± 1.1 fl (n = 24), respectively; P > 0.9, based on the Mann–Whitney test]. From these measurements, we can calculate very accurately the duration of the G1 phase (TG1) from the formula: TG1 = [ln(Critical size/Birth size)]/rate of size increase, as we described previously (Hoose et al. 2012; Soma et al. 2014). The length of the G1 phase was similar between WT (TG1 = 121 min) and uORFm-ACC1, FAS1 (TG1 = 115 min). Note that these estimates were from daughter cells, which have a longer cell cycle than mother S. cerevisiae cells.
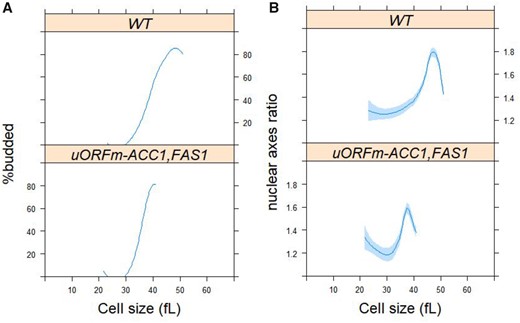
Derepressing translation of ACC1 and FAS1 promotes nuclear division, while inhibiting lipogenesis delays it. (A) Synchronous cultures of the indicated strains and conditions were obtained by elutriation (see Materials and Methods), from which the percentage of budded cells (y-axis) is shown against the mean cell size (in femtoliter; x-axis). (B) From the same samples as in (A), cells were processed for fluorescence microscopy to visualize the nucleus, as described in Materials and Methods (see also Supplementary Figure S4). Nuclear shape (y-axis) is shown against the mean cell size (in femtoliter; x-axis), from >2,000 cells/strain. Loess curves and the std errors at a 0.95 level are shown. The values used to generate the graphs are in Supplementary File S6/Sheet3.
Remarkably, compared to WT, uORFm-ACC1, FAS1 mutants initiated nuclear elongation and completed nuclear division disproportionately sooner—i.e., at a cell size some 10 fl smaller relative to WT cells (Figure 3B). For example, WT cells have a critical size of 40.6 fl (when half of them have budded; see above) and have to grow to about 47 fl until their nucleus is maximally elongated (see Figure 3B, top panel). In contrast, uORFm-ACC1, FAS1 mutants have a critical size of 36.7 fl (see above), but their nucleus reaches its maximal elongation only at about 38 fl (see Figure 3B, bottom panel). Taking into account, the rate of size increase in each strain (see above), once WT cells reach their critical site, their nucleus is maximally elongated about 27 min later. On the other hand, it takes only about 8 min for uORFm-ACC1, FAS1 mutants to maximally elongate their nucleus after they reach their critical size and pass through Start. Hence, the very pronounced acceleration of nuclear division in uORFm-ACC1, FAS1 cells is not the result of their very slight acceleration of Start, revealing a profound, active role of lipogenic enzymes late in the cell cycle.
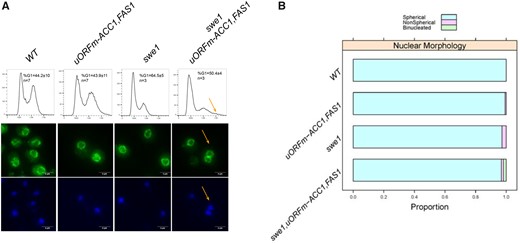
Loss of Swe1p in uORFm-ACC1, FAS1 cells leads to premature division. (A) Top, DNA content profiles of the indicated strains, obtained by flow cytometry. The x-axis on each histogram shows fluorescence per cell, and the y-axis the number of cells analyzed in each case. The average percentage of cells with G1 DNA content, standard deviation and number of independent experiments performed for each strain are shown. There was no statistically significant difference between WT and uORFm-ACC1, FAS1 cells (P = 0.96, based on the Mann–Whitney test). Representative fluorescent microscopy images of cells from the indicated strains, stained with an α-Nsp1 antibody (middle panels) or DAPI (bottom panels). The yellow arrows indicate a binucleate cell (middle and bottom panels) and cells with increased ploidy (top panel). (B) Summary of the proportion of cells with spherical, or not, nuclei from the strains shown and the proportion of cells with two nuclei, calculated from three independent experiments (from A).
We note that the doubling time of asynchronous cultures was the same for both WT and uORFm-ACC1, FAS1 cells (93 ± 2 m vs 91 ± 5 m, P = 0.3, based on the Mann–Whitney test, from at least four independent experiments in each case). Hence, although uORFm-ACC1, FAS1 cells complete nuclear division earlier than WT cells, the mutant cells appear to delay the completion of cytokinesis and cell separation. Together, these results reveal an active role for lipid synthesis in promoting nuclear division.
To further test that an increase in lipogenesis promotes nuclear division, we also examined nuclear morphology in asynchronous cells. Our analysis from synchronous cultures argued that uORFm-ACC1, FAS1 cells complete nuclear division earlier than WT cells but are then delayed in the completion of cytokinesis and cell separation. If so, in asynchronous cultures, the accelerated nuclear division should manifest in a lower frequency of cells displaying a mitotic nucleus, elongated across the bud neck, between the mother cell and the large bud. Conversely, suppose uORFm-ACC1, FAS1 cells are delayed in cytokinesis and cell separation. In that case, there should be more cells with a telophase nuclear morphology, with two separate nuclei, one in the mother cell and one in the large bud. Both of these predictions (i.e., a lower frequency of cells with elongated nuclei, but a higher frequency of cells with fully divided nuclei) were observed in uORFm-ACC1, FAS1 (n = 618 cells) compared to WT (n = 1037 cells) asynchronous cultures (see Supplementary Figure S5). The difference in the frequency distributions was highly significant (P < 0.0001; based on the chi-square test; χ2=60.35, one degree of freedom; see Supplementary Figure S5). Together with the kinetics of nuclear division in daughter cell synchronous cultures (Figure 3), these results provide strong support for the active role of lipogenesis in promoting nuclear division.
Increased lipogenesis promotes nuclear division independently of Swe1p
To place the accelerated nuclear division of the uORFm-ACC1, FAS1 cells in a functional pathway, we also examined cells lacking the Swe1p tyrosine kinase. Swe1p phosphorylates Cdc28p on Y19 and inhibits its mitotic kinase activity (Booher et al. 1993). Swe1p also drives the synthesis of sphingolipids (Chauhan et al. 2016). It has been reported that without Swe1p, cells enter mitosis prematurely and exit it at an abnormally small daughter birth size (Harvey and Kellogg 2003). We confirmed these phenotypes of swe1Δ cells. The birth size of swe1Δ cells was smaller than WT cells (17.2 ± 1.1 vs 22.3 ± 0.4 fl), from experiment-matched samples. Furthermore, swe1Δ cells had a smaller critical size than WT cells (35.8 ± 0.4 vs 40.6 ± 2 fl; see Supplementary Figure S6A), but the rate at which they increased in size was comparable to the rate for WT cells (0.31 vs 0.32/h, respectively). Similar to the accelerated initiation of nuclear division of uORFm-ACC1, FAS1 cells, swe1Δ cells also elongate their nuclear envelope at a significantly smaller size than WT cells (at 39 vs 48 fl, respectively; see Supplementary Figure S6B), consistent with the observations of Harvey and Kellogg (Harvey and Kellogg 2003). We note that normalized against their corresponding critical size, nuclear division is more disproportionately accelerated in uORFm-ACC1, FAS1 cells than in swe1Δ cells. But whereas swe1Δ cells go on to give birth to abnormally small cells, that is not the case for uORFm-ACC1, FAS1 cells, which are delayed in cytokinesis and generate daughters with normal size.
To see if the Swe1p-dependent mitotic acceleration functionally interacts with the translational derepression of lipogenesis, we deleted Swe1p in uORFm-ACC1, FAS1 cells. These mutants were viable, but they were not amenable to elutriation, and we could not query them in synchronous cultures. From asynchronous cultures, however, we found that the already small birth size of swe1Δ cells (17.2 ± 1.1 fl) was reduced even further (14.3 ± 0.3 fl) in swe1Δ, uORFm-ACC1, FAS1 cells. Furthermore, although the nucleus maintained its overall spherical shape (Figure 4, A and B), a small population of swe1Δ, uORFm-ACC1, FAS1 cells had increased ploidy. We reached this conclusion from DNA content profiles (Figure 4A, top panels) and nuclear staining (Figure 4A, middle and bottom panels). These results suggest that the accelerated nuclear division in uORFm-ACC1, FAS1 cells is largely additive to and independent of that observed in swe1Δ cells, with cells of the combined mutants born with an increased chance of undergoing aberrant mitosis.
Derepressing translation of ACC1 and FAS1 corrects mitotic phenotypes of sec14 mutants
We then turned our attention to other pathways that could functionally interact with the mitotic roles of the increased lipogenesis in uORFm-ACC1, FAS1 cells. Conditional sec14 mutants proliferate at 25°C but not at 37°C (Novick et al. 1980). Furthermore, inhibition of lipogenesis by cerulenin exacerbates the temperature sensitivity of sec14-1 cells (Dacquay et al. 2017). Whereas the cerulenin effect is a rather nonspecific phenotype (Lee et al. 2014), this observation raised the question of whether the increased lipogenesis in uORFm-ACC1, FAS1 cells could suppress the temperature-sensitive proliferation of sec14-1 cells. Indeed, coordinate ACC1 and FAS1 uORF inactivation partially rescued the growth of sec14-1 cells at the semipermissive temperature of 34°C (Figure 5A). This result suggests that increased lipogenesis reduced the threshold requirement of yeast cells for Sec14p. We also noted the appearance of a few suppressor colonies with robust growth at 34°C (Figure 5A).
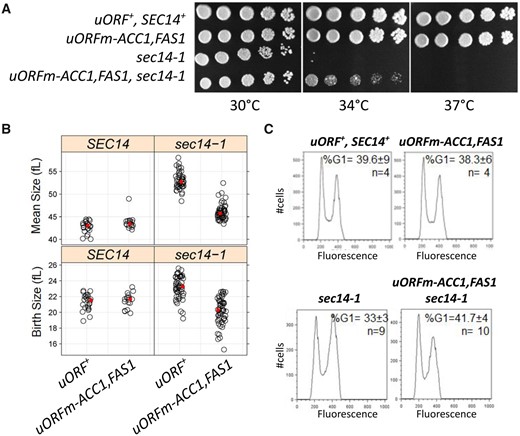
Derepressing the translational control of ACC1 and FAS1 suppresses the temperature sensitivity (A), large size (B), and high G2/M DNA content (C) of sec14-1 cells. (A) Serial dilutions (fivefold) of cultures of the indicated strains were spotted on solid media and grown at the temperatures shown. (B) Strip plots showing the mean (top panels) and birth (bottom panels) size (y-axis) for the indicated strains. The average in each case is in red. (C) Flow cytometry histograms for the indicated strains, with cell number on the y-axis and fluorescence per cell on the x-axis.
Although the Sec14p requirement for membrane trafficking through the yeast TGN/endosomal system is well-established (Bankaitis et al. 1989; Cleves et al. 1989; Nile et al. 2014), this PITP also executes cell-cycle functions. Even at the permissive temperature of 30°C sec14-1 cells exhibit larger birth and mean sizes and are delayed in passage through the G2/M stages of the cell cycle. This latter deficiency is manifested in asynchronous cultures as increased fractions of cells in G2/M (Huang et al. 2018). Also, sec14-1 mutants must reach a larger critical size before initiating cell division (Huang et al. 2018). Strikingly, whereas ACC1 and FAS1 uORF mutations did not alter WT cells’ size, these uORF mutations corrected to a significant extent the larger birth and mean size of sec14-1 cells (Figure 5B, right panels; P > 0.05, Kruskal–Wallis, and post hoc Nemenyi tests). Moreover, the combined uORF mutations fully corrected the G2/M delay characteristic of sec14-1ts cells (Figure 5C). These collective results cannot be accounted for by simple models where cell enlargement is accommodated by new plasma membrane synthesis before division.
Sec14p has not been previously implicated in nuclear division. But since our data suggest that ACC1 and FAS1 uORF mutations correct the various cell cycle-related phenotypes of sec14-1 cells, we examined the timing of nuclear division in synchronized cultures produced by elutriation. As we had previously reported (Huang et al. 2018), the critical size of sec14-1 cells is larger than that of WT cells even at the permissive temperature of 30°C (Figure 6A, compare the top two panels). Furthermore, sec14-1 cells initiated and completed nuclear division at a significantly larger size relative to WT cells (Figure 6B). Strikingly, ACC1 and FAS1 uORF mutations corrected both the larger critical size and the delayed nuclear division of sec14-1 cells (Figure 6, A and B). However, the scheduling of nuclear division in uORFm-ACC1, FAS1, sec14-1 mutants was similar to that of WT cells and was not accelerated as observed in uORFm-ACC1, FAS1 mutants (Figure 3B). These collective data strongly support the idea that increased lipogenesis provides for the mitotic functions of Sec14p and suggests that the accelerated nuclear division schedule associated with elevated lipogenesis still requires some Sec14p involvement.
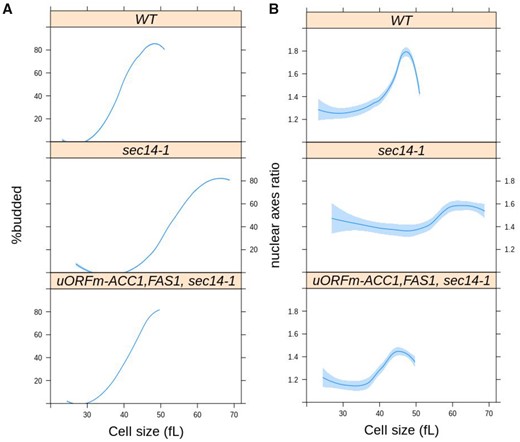
Loss-of-function sec14-1 mutants delay nuclear division, but they are suppressed by the uORF mutations in ACC1 and FAS1. From synchronous cultures of the indicated strains, the percentage of budded cells (A) and nuclear shape (B) as a function of cell size (x-axis) were measured and plotted as in Figure 3. For the WT cells, the values were the same as those in Figure 3. The values used to generate the graphs are in Supplementary File S6/Sheet5.
Specificity of “bypass Sec14p” mechanisms in correcting the delayed nuclear division of sec14-1 mutants
The ACC1 and FAS1 uORF mutations perturb lipid metabolism (Figure 2). But fatty acids are the precursors for all glycerolipids and sphingolipids. Hence, it is difficult to reduce to a manageable level the possibilities regarding which parts of lipid metabolism are primarily responsible for disturbed nuclear morphogenesis. Since our data point to functional interactions between the increased lipogenesis in uORFm-ACC1, FAS1 cells, and Sec14p-dependent processes (Figures 5 and 6), the known roles of Sec14p were exploited as a tool that reports on distinct arms of the lipid metabolome.
As a PtdIns/PtdCho transfer protein (Bankaitis et al. 1990; Grabon et al. 2019), Sec14p leverages its heterotypic lipid binding and exchange activities to potentiate the activities of PtdIns 4-OH kinases and thereby stimulate PtdIns-4-phosphate signaling (Schaaf et al. 2008; Bankaitis et al. 2010). It is by this mechanism that Sec14p and other PITPs function as sensors of lipid metabolism and integrate the activities of distinct arms of the lipid metabolome with localized outputs of PtdIns-4-phosphate signaling (Wang et al. 2019). In that regard, Sec14p executes essential cellular activities, but the normally essential Sec14p requirement for cell viability is bypassed by derangements in specific aspects of lipid metabolism. These include loss-of-function mutations in the PtdIns-4-phosphate phosphatase Sac1p (Cleves et al. 1989; Rivas et al. 1999), the CDP-choline pathway (but not the de novo pathway) for PtdCho biosynthesis (Cleves et al. 1991; Xie et al. 2001), and the ergosterol and PtdIns-4-phosphate exchange protein Kes1p (Fang et al. 1996; Li et al. 2002; Mousley et al. 2012).
With regard to bypass Sec14p mutations in the CDP-choline pathway, the lethality and secretory defects of sec14-1 mutants at 37°C are suppressed by loss of the Cki1p choline kinase which catalyzes the first reaction of this PtdCho biosynthetic pathway (Cleves et al. 1991). However, even though genetic ablation of Cki1p activity yielded cells that exhibited a slightly smaller critical size (consistent with slightly accelerated initiation of cell division), this metabolic defect failed to suppress the delayed nuclear division of sec14-1 mutants (Figure 7). Strikingly different results were obtained for Kes1p loss-of-function mutations which we previously demonstrated to correct the large cell size and cell cycle delay of sec14-1 cells [Huang et al. (2018); see also Figure 7A]. Kes1p inactivation fully corrected the delay in the nuclear division of sec14-1 cells (Figure 7B). These results indicate a sharp differentiation between “bypass Sec14p” mutants in their abilities to correct the derangements in nuclear division associated with Sec14p dysfunction, even though these “bypass Sec14p” mutations share in common their abilities to restore cell viability and efficient membrane trafficking to cells ablated for Sec14p function. These data also indicate that defects in the CDP-choline pathway of PtdCho synthesis do not significantly impact nuclear division.
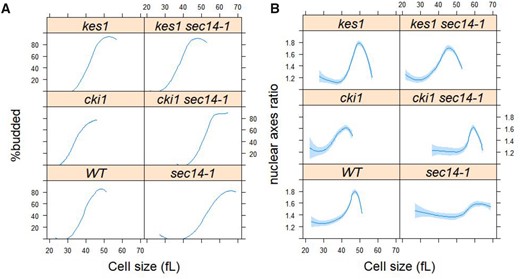
Loss of Kes1p, but not Cki1p, suppresses the delayed nuclear division of sec14-1 mutants. From synchronous cultures of the indicated strains, the percentage of budded cells (A) and nuclear shape (B) as a function of cell size (x-axis) were measured and plotted as in Figure 6. For the wild type cells (WT) and sec14-1 cells, the values were the same as those in Figure 6. The values used to generate the graphs are in Supplementary File S6/Sheet6.
Enhanced ACC1 and FAS1 translation corrects the aberrant nuclear shape of PtdIns 4-OH kinase mutants
A major function of Sec14p in cells is to stimulate the activities of the Golgi/endosomal and plasma membrane PtdIns 4-OH kinases Pik1p and Stt4p, respectively (Schaaf et al. 2008; Bankaitis et al. 2010). We, therefore, interrogated the cell cycle and nuclear morphologic phenotypes of temperature-sensitive mutants defective in the activities of these two lipid kinases as each enzyme is individually essential for yeast cell viability, and these two enzymes together account for nearly all of the PtdIns 4-OH kinase activity in cells (Audhya et al. 2000). Even at their permissive temperature (25°C), pik1-101 and stt4-4 cells grew slowly and exhibited aberrant cell morphologies. Indeed, we were unable to obtain synchronous cultures of these mutants by elutriation. In an alternative approach, we examined asynchronous cultures of pik1-101 and stt4-4 cells for cell cycle parameters, such as cell size and DNA content. At the permissive temperature of 25°C, both pik1-101 and stt4-4 cells were significantly larger than WT cells (∼50 and 25%, respectively; Supplementary Figure S7A). Moreover, the DNA contents of both mutants were also irregular (Supplementary Figure S7B). Although Pik1p inactivation results in a cytokinesis defect at 37°C (Garcia-Bustos et al. 1994), we did not obtain evidence of increased ploidy at 25°C. Rather, the DNA content profiles for both mutants consistently reported a substantial delay in passage through the S phase (Supplementary Figure S7B).
The cell cycle phenotypes described above, when coupled with prior evidence that phospholipid and triacylglycerol synthesis impacts nuclear morphology (Webster et al. 2010; Siniossoglou 2013; Barbosa et al. 2019), prompted us to examine the nuclear morphologies of pik1-101 and stt4-4 cells. These mutants exhibited morphologically abnormal nuclei characterized by highly irregular shapes, especially for pik1-101 cells and to a lesser extent for stt4-4 cells (Figure 8). Remarkably, uORFm-ACC1, FAS1 restored normal nuclear morphology to pik1-101 and stt4-4 mutants (Figure 8). In stt4-4 cells, whose nuclear morphology was not as severely perturbed, the suppressive effect of increased lipogenesis was not statistically significant (P = 0.07, based on the Kruskal–Wallis and post hoc Nemenyi tests; see also Supplementary Figure S8). In pik1-101 cells, increased lipogenesis levied a significant (P = 0.01) correction of the abnormal nuclear shape of PtdIns 4-OH kinase mutants. These results further support the notion that lipogenesis actively controls nuclear shape and division. They also identify a role for PtdIns-4-phosphate signaling in homeostatic control of nuclear morphology.
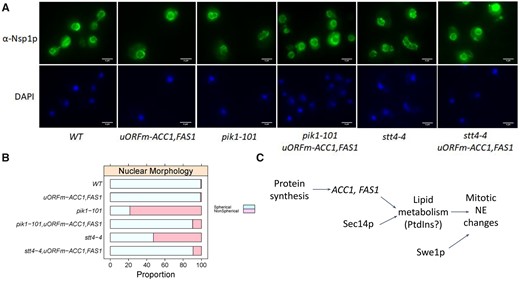
Upregulating translation of ACC1 and FAS1 corrects the aberrant nuclear shape of PI-4-kinase mutants. (A) Representative fluorescent microscopy images of cells from the indicated strains, stained with an α-Nsp1 antibody (top panels) or DAPI (bottom panels). (B) Summary of the proportion of cells with spherical, or not, nuclei from the strains shown, calculated from three independent experiments (see Supplementary Figure S8). (C) Schematic summary of our results.
Because Pik1p is in the cytoplasm and the nucleus (Strahl et al. 2005), we also examined if nuclear or cytoplasmic PtdIns-4-phosphate may be preferentially involved in nuclear homeostasis. We queried nuclear morphology of pik1-101 cells carrying a plasmid that drives expression of an active but truncated Pik1p, which is unable to exit the nucleus [Pik1p(Δ10-192), because its nuclear export signal is deleted] (Strahl et al. 2005). Pik1p(Δ10-192) could not restore the nuclear shape of pik1-101 cells to a spherical one (see Supplementary Figure S9). These data suggest that generation of cytoplasmic PtdIns-4-phosphate is necessary for maintaining the spherical shape of the nucleus.
Discussion
Our results suggest that gain-of-function mutations affecting translational control of lipogenic enzymes accelerate nuclear division, a critical cell cycle process. To our knowledge, these results provide the first example of a metabolic pathway that is not merely required for a cell cycle process but actively drives the process forward. These findings are discussed in the context of cell cycle-dependent translational control of lipogenesis as it relates to the regulation of nuclear morphogenesis.
Translational control of lipogenic enzymes
The question of how mammalian acetyl-CoA carboxylase (ACC1, ACACA) and fatty acid synthase (FASN) are regulated is of intense interest because of the central role these enzymes play in the control of food intake, their contributions to obesity, and their association with cancer (Kuhajda et al. 1994; Baenke et al. 2013; Beloribi-Djefaflia et al. 2016). ACC1 expression is increased in conjunction with FASN to upregulate lipid synthesis in human tumors. Multiple mechanisms have been associated with the levels and activity of these enzymes in mammals. The activities of these enzymes in mammals are subject to various layers of regulation, and the control of Acc1 activity by phosphorylation has garnered considerable attention (Kim 1997). Acc1 is inactivated upon phosphorylation by AMPK and, as leptin activates AMPK, this regulation is a crucial aspect of the mechanism by which leptin suppresses appetite (Gao et al. 2007). The AMPK-mediated inhibition of Acc1 is relieved just before cytokinesis, and this timing correlates with increased Acc1 enzymatic activity late in the Hela cell cycle (Scaglia et al. 2014). However, the precise role of phosphorylation in the cell cycle regulation of Acc1p remains unclear. Large-scale phosphoproteomic analyses identify several phosphorylation sites on yeast Acc1p. The major site is at residue S1157, which corresponds to S1216 in the human enzyme (Hunkeler et al. 2016). Although this site is conserved in the Acc1p enzymes of all eukaryotes, there are no reports that Acc1pS1216 phosphorylation is of any regulatory consequence for mammalian Acc1p. Moreover, there is no evidence that Acc1p phosphorylation is subject to cell cycle control in yeast.
Our data identify translation control as a mechanism for regulating lipogenesis as a function of the cell cycle. We had previously shown that translation of the mRNAs encoding Acc1p and Fas1,2p peaks late in the cell cycle (Blank et al. 2017a; see also Supplementary Figure S2). That this represents a broadly conserved layer of regulation is indicated by subsequent studies that showed translational control of human ACC1 is required for the progression of quiescent T cells into a proliferative state (Ricciardi et al. 2018). These prior studies linking elevated Acc1p and Fas1p activity with increased cell proliferation established these enzymes are necessary for lipogenesis and cell division. In that regard, there are numerous other examples where metabolic and cell growth processes are required for various cell cycle events (Johnston et al. 1977).
The novelty of the current study rests on our demonstration that upregulating Acc1p and Fas1p (by abrogating translational inhibitory elements) is sufficient to drive a critical cell cycle process—i.e., nuclear division. Functional ablation of the inhibitory uORFs that lie upstream of each of the corresponding structural genes elevated the overall levels of these enzymes while preserving the cell cycle-dependent oscillations in their abundance, albeit with a dampened amplitude (Blank et al. 2017a; see also Supplementary Figure S2). This is an expected result because, for the uORF-mediated control to fully account for the mitotic peak of translation of ACC1 and FAS1, the cellular ribosome content must also fluctuate as cells progress through the cell cycle. Evidence from our laboratory and others argues against such cell cycle fluctuations in ribosome content (Shulman et al. 1973; Elliott et al. 1979; Blank et al. 2020). Whole-cell metabolic profiling demonstrates lipid abundance (including all major classes; PtdIns, PtdCho, PtdEtn, and TG) is the most periodic metabolic parameter as a function of the cell cycle and that it peaks in mitosis (Blank et al. 2020). Furthermore, we now show that enhanced ACC1 and FAS1 translation was sufficient to increase most lipids’ abundances (Figure 2).
Nuclear morphology and lipogenesis
Yeast and other fungi undergo closed mitosis, where the nuclear envelope remains intact throughout the entire cell cycle. The nucleus maintains a spherical shape throughout interphase and then elongates during mitosis (Meseroll and Cohen-Fix 2016). The fact that nuclear envelope expansion depends on cell cycle progression and not on a general increase in cell size argues for specialized metabolic inputs, including fatty acid and phospholipid synthesis (Walters et al. 2019). Indeed, nuclear membrane proliferation is required for progression through mitosis, regardless of whether it is closed or disassembled before reassembling during telophase as in mammalian cells. Inhibition of FAS impairs nuclear elongation and chromosome segregation in fission yeast (Takemoto et al. 2016). A mitotic delay is occasionally accompanied by the appearance of nuclear membrane extensions, or flares, in budding yeast. These flares are interpreted as evidence for continued nuclear elongation during some cell cycle arrest (Walters et al. 2012; Meseroll and Cohen-Fix 2016). In support of this interpretation, ACC1, FAS1, or FAS2 loss-of-function mutations impair flare formation and nuclear elongation (Walters et al. 2014). However, we emphasize that the acceleration of nuclear elongation and division that we observed in the face of the upregulated translation of lipogenic enzymes is without precedent. Those data are not only fully consistent with the requirement for lipogenesis in nuclear elongation and division, but these also establish that enhanced lipogenesis is sufficient to drive these processes.
Phosphoinositide signaling in control of nuclear division and morphology
While the role of the normally essential Sec14p in membrane trafficking is well documented (Bankaitis et al. 1989; Cleves et al. 1989; Fang et al. 1996), there is evidence that the essential cellular function(s) executed by Sec14p are not solely at the level of membrane trafficking control but also at the level of cell cycle control (Mousley et al. 2012). In that regard, Sec14p hypomorphisms manifest themselves in cell cycle phenotypes such as delay in the passage through G2/M and progression from G1 into S-phase (Huang et al. 2018). In that context, we report an unexpected confluence of translational control of lipogenesis with Sec14p-dependent phosphoinositide signaling in the cell cycle. We find that sec14-1 mutants are not only delayed in the initiation and completion of nuclear division but that gain-of-function mutations that ablate the ACC1 and FAS1 uORFs (with the result that the two corresponding mRNAs are translationally derepressed) partially rescue sec14-1 growth defects. Moreover, ablation of the uORFs fully rescued the delays in progression through G2/M and G1/S in Sec14p-deficient mutants. What is particularly striking in light of those data is that the accelerated timing of nuclear division in the uORF mutants was corrected in the sec14-1ts genetic background. These collective data not only indicate that increased lipogenesis suppresses the mitotic functions of Sec14p but also suggest that the accelerated nuclear division schedule associated with elevated lipogenesis requires Sec14p involvement.
The available evidence indicates that the Sec14p involvement was independent of its role in membrane trafficking on two counts. First, these phenotypes were manifested at permissive temperatures for sec14-1 mutants where membrane trafficking pathways are operating normally by all measurable criteria. Second, the “bypass Sec14” mutant that inactivates Cki1p (i.e., the choline kinase representing the first step in the CDP-choline pathway for PtdCho biosynthesis) corrects the membrane trafficking defects of sec14 mutants but fails to correct the deranged scheduling of nuclear elongation and division in sec14-1 mutants. Rather, as Sec14p stimulates PtdIns-4-P synthesis by both the Pik1p and Stt4p kinases in vivo, the data argue that Sec14p-mediated phosphoinositide signaling is involved in regulating nuclear elongation and division. Two lines of evidence support this interpretation of the data. First, inactivation of Kes1p, an antagonist of Sec14p- and Pik1p-dependent PtdIns-4-P signaling (Fang et al. 1996; Li et al. 2002), corrects both the membrane trafficking and the nuclear elongation/division defects associated with Sec14p deficiencies. In this manner, the kes1 “bypass Sec14p” mutants differ from cki1 mutants that correct only the membrane trafficking deficits. Second, cells compromised for Pik1p activity presented misshapen nuclei whose morphological derangements were corrected by increased lipogenesis. These data are intriguing in light of previous demonstrations that Pik1p shuttles between cytosolic and nuclear pools and executes essential cellular functions in both compartments (Garcia-Bustos et al. 1994; Strahl et al. 2005). Our data argue strongly that cytoplasmic Pik1p is necessary for its role in nuclear division (see Supplementary Figure S9).
Precisely how PtdIns-4-P signaling modulates nuclear division remains to be determined. One execution point could be at the translational control level as inactivation of Pik1p or Stt4p triggers a block in protein synthesis through phosphorylation of translation initiation factor eIF2ɑ (Cameroni et al. 2006). Another attractive and not mutually exclusive possibility is that PtdIns-4-P serves as a precursor to synthesizing a nuclear pool of PtdIns-4,5-bisphosphate (PtdIns-4,5-P2). There is abundant literature regarding nuclear PtdIns-4,5-P2 and its roles (direct and indirect) in regulating multiple nuclear events such as transcription, mRNA processing and polyadenylation, and mRNA export (Cocco et al. 1987, 1989; York et al. 1999; Li et al. 2013; Hamann and Blind 2018; Chen et al. 2020). We attempted to assess the relationship between PtdIns-4,5-P2 synthesis and ACC1 and FAS1 translational control but were unsuccessful in generating the requisite strains for the analysis. So, the relationship between PtdIns-4-P and PtdIns-4,5-P2 signaling related to nuclear elongation and division remains to be resolved.
In summary, the results reported here emphasize the physiological significance of generating specific lipid pools that drive a crucial cell cycle process—i.e., nuclear elongation and division. These lipids pools represent metabolic inputs that do not merely “fuel” a cell cycle process but, rather, play heretofore unappreciated active and instructive signaling roles in driving landmark cell cycle events. The collective data integrate protein synthesis, lipogenesis, and phosphoinositide signaling with the timing of nuclear elongation and division. In this manner, this study sets the stage for deciphering the specific identities and compartmental organization of the relevant lipid pools that control these processes.
Data availability
Strains and plasmids are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, and tables.
The supplementary material is available through figshare: https://doi.org/10.6084/m9.figshare.16649341.
Acknowledgments
We thank Jeremy Thorner (UC Berkeley) for the pTS13 [PGAL1-Pik1p(Δ10-192)] plasmid.
Funding
This work was supported by National Institutes of Health grant R01 GM123139 to M.P., while V.A.B. was supported by NIH grant R35 GM131804.
Conflicts of interest
The authors declare that there is no conflict of interest.
Literature cited
Author notes
Present address: Institute of Biosciences and Technology, Texas A&M University, Houston, TX 77843, USA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}