





Abstract
Cytochrome c oxidase (CcO) is a multimeric copper-containing enzyme of the mitochondrial respiratory chain that powers cellular energy production. The two core subunits of cytochrome c oxidase, Cox1 and Cox2, harbor the catalytic CuB and CuA sites, respectively. Biogenesis of each copper site occurs separately and requires multiple proteins that constitute the mitochondrial copper delivery pathway. Currently, the identity of all the members of the pathway is not known, though several evolutionarily conserved twin CX9C motif-containing proteins have been implicated in this process. Here, we performed a targeted yeast suppressor screen that placed Coa4, a twin CX9C motif-containing protein, in the copper delivery pathway to the Cox1 subunit. Specifically, we show that overexpression of Cox11, a copper metallochaperone required for the formation of CuB site, can restore Cox1 abundance, cytochrome c oxidase assembly, and mitochondrial respiration in coa4Δ cells. This rescue is dependent on the copper-coordinating cysteines of Cox11. The abundance of Coa4 and Cox11 in mitochondria is reciprocally regulated, further linking Coa4 to the CuB site biogenesis. Additionally, we find that coa4Δ cells have reduced levels of copper and exogenous copper supplementation can partially ameliorate its respiratory-deficient phenotype, a finding that connects Coa4 to cellular copper homeostasis. Finally, we demonstrate that human COA4 can replace the function of yeast Coa4 indicating its evolutionarily conserved role. Our work provides genetic evidences for the role of Coa4 in the copper delivery pathway to the CuB site of cytochrome c oxidase.
Introduction
Cytochrome c oxidase (CcO) is the terminal enzyme of the mitochondrial respiratory chain that catalyzes the transfer of electrons from cytochrome c to molecular oxygen (Gennis and Ferguson-Miller 1995; Ferguson-Miller and Babcock 1996; Yoshikawa and Shimada 2015; Guo et al. 2018). It is a large, membrane-bound multimeric protein complex composed of 3 core subunits encoded by the mitochondrial DNA (mtDNA) and additional (9–11) subunits encoded by the nuclear DNA (Zong et al. 2018; Hartley et al. 2019). In addition to these subunits, CcO contains copper and heme as redox-active cofactors essential for its activity and stability (Gennis and Ferguson-Miller 1995; Ferguson-Miller and Babcock 1996). Copper and heme are coordinated to the two core subunits of CcO—Cox1 and Cox2—forming the essential CuA, heme a, and heme a3/CuB redox sites that are directly involved in the electron transfer to molecular oxygen (Tsukihara et al. 1995, 1996).
Biogenesis of the CcO holoenzyme is a particularly complex process requiring almost 30 assembly factors that are involved in diverse processes including the expression of mtDNA-encoded subunits, their insertion into the membrane, metal trafficking, cofactor biosynthesis and insertion, and stability of the assembly intermediates (Timon-Gomez et al. 2018). Loss-of-function mutations in many of these assembly factors have been identified in patients with a wide range of clinical pathologies including Leigh syndrome, hypertrophic cardiomyopathy, and ketoacidotic coma (Jaksch et al. 2000; Valnot et al. 2000; Péquignot et al. 2001; Shoubridge 2001; Huigsloot et al. 2011; Lee et al. 2012; Baertling et al. 2015). Currently, functions of many of these CcO assembly factors are unknown, which impedes our understanding of CcO biogenesis and developing therapeutic approaches for these devastating disorders.
The first step in defining the function of CcO assembly factors is to link them to the above-described cellular processes in CcO biogenesis. These processes are spatially separated in the mitochondria, with factors required for translation of mtDNA-encoded subunits present in the mitochondrial matrix, factors required for membrane insertion present in the inner mitochondrial membrane, and factors required for copper delivery present in the intermembrane space (IMS) of mitochondria (Timon-Gomez et al. 2018). Copper delivery to Cox1 and Cox2 in the IMS occurs in a modular fashion, where the assembly of each module requires a dedicated set of evolutionarily conserved proteins that constitute the mitochondrial copper delivery pathway (Fig. 1). Prior studies have established that IMS-localized Cox17 is the common source of copper for both Cox1- and Cox2-specific metallochaperones, Cox11 and Sco1, respectively (Fig. 1;Carr et al. 2002; Horng et al. 2004; Morgada et al. 2015). These metallochaperones require redox proteins including Sco2 and Coa6 for the biogenesis of CuA site (Fig. 1; Morgada et al. 2015; Soma et al. 2019). In addition to these proteins, other twin CX9C motif-containing IMS proteins including Cox19, Cox23, Cmc2, Pet191, and Coa4 have been implicated in the assembly of CcO (Longen et al. 2009). However, it is not known whether they are part of the copper delivery pathway and if so, which module are they required for and where precisely they are placed in the pathway.
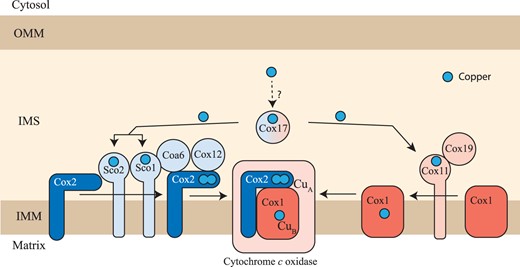
The mitochondrial copper delivery pathway to cytochrome c oxidase. A schematic diagram of the mitochondrial copper delivery pathway to CcO is shown. The assembly of the Cox1 and Cox2 subunits of CcO is a modular process requiring a dedicated set of assembly factors that are specific for each module. Cox11 and Cox19 are involved in CuB site formation within the Cox1 subunit whereas Sco1, Sco2, Cox12, and Coa6 are involved in the formation of CuA site within the Cox2 subunit. Cox11 is the specific copper metallochaperone for the Cox1 subunit and Sco1 is the specific copper metallochaperone for the Cox2 subunit. Cox11 and Sco proteins receive their copper from Cox17, a twin CX9C motif-containing IMS protein. Sco2 and Coa6 function as disulfide reductases in the copper transfer from Sco1 to Cox2 while Cox19 is required to maintain Cox11 in a functional form. OMM, outer mitochondrial membrane; IMS, intermembrane space; IMM, inner mitochondrial membrane.
To this end, we performed a targeted genetic suppressor screen using yeast mutants of twin CX9C or its variant CX9C—CX10C proteins (which we refer to as CX9CXnCX10C) and uncovered a novel genetic interaction between Cox11 and Coa4, a CcO assembly factor with unknown function. Our follow-up biochemical and genetic studies placed Coa4 in the copper delivery pathway to the CuB site of CcO. We further showed that coa4Δ cells have reduced cellular copper levels, which contributes to its respiratory growth defect, a phenotype that can be partially rescued by copper supplementation. Our study uncovers an evolutionarily conserved role of Coa4 in mitochondrial copper biology and places it in the mitochondrial copper delivery pathway to the Cox1 subunit of CcO.
Materials and methods
Yeast strains and growth conditions
The yeast Saccharomyces cerevisiae mutants used in this study are listed in Table 1. The yeast strains were confirmed by polymerase chain reaction (PCR)-based genotyping. Yeast cells were cultured in either YPD (1% yeast extract, 2% peptone, and 2% dextrose), YPGE (1% yeast extract, 2% peptone, 3% glycerol, and 1% ethanol) or YPGal (1% yeast extract, 2% peptone, and 2% galactose) media. YPD and YPGE plates were prepared by the addition of 2% agar. For copper supplementation experiments, 100 mM stock solution of CuCl2 was prepared and filter sterilized and was added to the autoclaved growth media at a final concentration of 10 μM. For growth on agar plates, 3 μL of 10x serial dilutions of precultures were seeded onto YPD or YPGE plates and incubated at 30°C or 37°C for the indicated period. For growth in liquid medium, yeast cells were precultured in YPD or in the appropriate synthetic dropout media containing glucose as the carbon source. The preculture was washed and resuspended in autoclaved water and was inoculated in the indicated media at a starting OD of 0.1. Growth in liquid media was monitored spectrophotometrically at 600 nm. For mitochondrial isolation, yeast cells were grown in the indicated media and were harvested at the late-log phase.
Yeast strains used in this study.
| Yeast strains | Genotype | Source |
|---|---|---|
| BY4741 WT | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4742 WT | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4743 WT | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2 | Khalimonchuk, O. |
| BY4741 cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox11Δ::KanMX4 | Open Biosystems |
| BY4741 cox12Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox12Δ::kanMX4 | Open Biosystems |
| BY4741 cox17Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox17Δ::kanMX4 | Open Biosystems |
| BY4741 cox19Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox19Δ::KanMX4 | Open Biosystems |
| BY4741 cox23Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox23Δ::kanMX4 | Open Biosystems |
| BY4741 sco1Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco1Δ::KanMX4 | Open Biosystems |
| BY4741 sco2Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco2Δ::KanMX4 | Open Biosystems |
| BY4741 cmc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc1Δ::kanMX4 | Open Biosystems |
| BY4741 cmc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc2Δ::URA3 | Barrientos, A. |
| BY4742 coa4Δ | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0, coa4Δ::hphNT1 | This study |
| BY4743 pet191Δ | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2, pet191Δ::kanMX4/pet191Δ::kanMX4 | Khalimonchuk O. |
| BY4741 coa6Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa6Δ::kanMX4 | Open Biosystems |
| STY10 coa6Δsco2Δ | MAT a, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0, sco2Δ::KanMX4, coa6Δ::NatMX4 | Ghosh et al., 2016 |
| BY4742 coa4Δ cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa4Δ::hphNT1, cox11Δ::natMX4 | This study |
| BY4742 coa4Δd | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, coa4Δ:: hphNT1 | This study |
| Yeast strains | Genotype | Source |
|---|---|---|
| BY4741 WT | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4742 WT | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4743 WT | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2 | Khalimonchuk, O. |
| BY4741 cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox11Δ::KanMX4 | Open Biosystems |
| BY4741 cox12Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox12Δ::kanMX4 | Open Biosystems |
| BY4741 cox17Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox17Δ::kanMX4 | Open Biosystems |
| BY4741 cox19Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox19Δ::KanMX4 | Open Biosystems |
| BY4741 cox23Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox23Δ::kanMX4 | Open Biosystems |
| BY4741 sco1Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco1Δ::KanMX4 | Open Biosystems |
| BY4741 sco2Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco2Δ::KanMX4 | Open Biosystems |
| BY4741 cmc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc1Δ::kanMX4 | Open Biosystems |
| BY4741 cmc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc2Δ::URA3 | Barrientos, A. |
| BY4742 coa4Δ | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0, coa4Δ::hphNT1 | This study |
| BY4743 pet191Δ | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2, pet191Δ::kanMX4/pet191Δ::kanMX4 | Khalimonchuk O. |
| BY4741 coa6Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa6Δ::kanMX4 | Open Biosystems |
| STY10 coa6Δsco2Δ | MAT a, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0, sco2Δ::KanMX4, coa6Δ::NatMX4 | Ghosh et al., 2016 |
| BY4742 coa4Δ cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa4Δ::hphNT1, cox11Δ::natMX4 | This study |
| BY4742 coa4Δd | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, coa4Δ:: hphNT1 | This study |
Yeast strains used in this study.
| Yeast strains | Genotype | Source |
|---|---|---|
| BY4741 WT | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4742 WT | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4743 WT | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2 | Khalimonchuk, O. |
| BY4741 cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox11Δ::KanMX4 | Open Biosystems |
| BY4741 cox12Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox12Δ::kanMX4 | Open Biosystems |
| BY4741 cox17Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox17Δ::kanMX4 | Open Biosystems |
| BY4741 cox19Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox19Δ::KanMX4 | Open Biosystems |
| BY4741 cox23Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox23Δ::kanMX4 | Open Biosystems |
| BY4741 sco1Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco1Δ::KanMX4 | Open Biosystems |
| BY4741 sco2Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco2Δ::KanMX4 | Open Biosystems |
| BY4741 cmc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc1Δ::kanMX4 | Open Biosystems |
| BY4741 cmc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc2Δ::URA3 | Barrientos, A. |
| BY4742 coa4Δ | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0, coa4Δ::hphNT1 | This study |
| BY4743 pet191Δ | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2, pet191Δ::kanMX4/pet191Δ::kanMX4 | Khalimonchuk O. |
| BY4741 coa6Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa6Δ::kanMX4 | Open Biosystems |
| STY10 coa6Δsco2Δ | MAT a, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0, sco2Δ::KanMX4, coa6Δ::NatMX4 | Ghosh et al., 2016 |
| BY4742 coa4Δ cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa4Δ::hphNT1, cox11Δ::natMX4 | This study |
| BY4742 coa4Δd | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, coa4Δ:: hphNT1 | This study |
| Yeast strains | Genotype | Source |
|---|---|---|
| BY4741 WT | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4742 WT | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0 | Greenberg, M.L. |
| BY4743 WT | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2 | Khalimonchuk, O. |
| BY4741 cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox11Δ::KanMX4 | Open Biosystems |
| BY4741 cox12Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox12Δ::kanMX4 | Open Biosystems |
| BY4741 cox17Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox17Δ::kanMX4 | Open Biosystems |
| BY4741 cox19Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox19Δ::KanMX4 | Open Biosystems |
| BY4741 cox23Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cox23Δ::kanMX4 | Open Biosystems |
| BY4741 sco1Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco1Δ::KanMX4 | Open Biosystems |
| BY4741 sco2Δ | MAT a, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, sco2Δ::KanMX4 | Open Biosystems |
| BY4741 cmc1Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc1Δ::kanMX4 | Open Biosystems |
| BY4741 cmc2Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, cmc2Δ::URA3 | Barrientos, A. |
| BY4742 coa4Δ | MATα, his 3Δ1, leu 2Δ0, lys2Δ0, ura3Δ0, coa4Δ::hphNT1 | This study |
| BY4743 pet191Δ | MATa/α his 3Δ1/his 3Δ1, leu2Δ0/leu2Δ0, ura3Δ0/ura3Δ0, met15Δ/MET15, lys2Δ0/LYS2, pet191Δ::kanMX4/pet191Δ::kanMX4 | Khalimonchuk O. |
| BY4741 coa6Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa6Δ::kanMX4 | Open Biosystems |
| STY10 coa6Δsco2Δ | MAT a, his3Δ1, leu2Δ0, ura3Δ0, lys2Δ0, sco2Δ::KanMX4, coa6Δ::NatMX4 | Ghosh et al., 2016 |
| BY4742 coa4Δ cox11Δ | MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0, coa4Δ::hphNT1, cox11Δ::natMX4 | This study |
| BY4742 coa4Δd | MATα, his3Δ1, leu2Δ0, lys2Δ0, ura3Δ0, coa4Δ:: hphNT1 | This study |
Plasmids
The primers used for gene amplification are listed in Supplementary Table 1. The coding sequence along with an upstream region of approximately 500–600 bp containing their putative promoter was cloned in the pRS416 plasmid at the multiple cloning site. The cloned fragments were verified by sequencing. pRS415 plasmids were constructed by subcloning the gene along with its promoter from pRS416 into the pRS415 empty vector. For cloning human COA4, a codon-optimized V5-tagged human COA4 sequence, commercially obtained from GenScript, was used as a template to amplify V5-tagged hsCOA4 (hsCOA4-V5) and untagged hsCOA4 (hsCOA4) using the primers listed in Supplementary Table 1. The PCR amplified sequence was inserted downstream of the putative promoter region of yeast COA4 in the pRS416 plasmid. Site-directed mutagenesis was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent #210518) using the primers listed in Supplementary Table 2.
Yeast transformation
Yeast transformation was performed as described before (Chen et al. 1992). Briefly, cells were grown in YPD overnight at 30°C and were washed and resuspended in one-step buffer [0.2 N Lithium acetate, 40% PEG 3350, and 100 mM dithiothreitol (DTT)] along with 1 µg of plasmid DNA and 50 µg of salmon sperm DNA. The mixture was incubated at room temperature for 25 min, 42°C for 20 min, followed by incubation on ice for 5 min. Cells were then pelleted, washed, and plated on appropriate synthetic dropout plates and incubated at 30°C for about 2–3 days to obtain colonies.
Construction of the coa4Δd strain
The COA4 gene was disrupted in the WT BY4742 background by replacing the codon for Cys39 (5′-TGC-3′) with a stop codon (5′-TGA-3′) and replacing the COA4 DNA sequence corresponding to amino acids 40–74 with the hygromycin (hygro) gene cassette. The hygro cassette was amplified from the pFA6-hphNT1 plasmid by using the following PCR primers: (1) 5′-GACCCGGATGTGTGGGACACGAGAAT ATCCAAGACCGGATGACGTACGCTGCAGGTCGAC-3′ and (2) 5′-GTCCCATCCACGTCC ACTGTGCTTACGCGCTCTCTATTACCATCGATGAATTCGAGCTCG-3′. The linear cassette was then transformed into WT BY4742 cells by high-efficiency transformation.
Cellular copper measurements
Cellular copper levels were measured using inductively coupled plasma mass spectrometry (ICP-MS; NexION 300D instrument from PerkinElmer Inc). Briefly, yeast cells were precultured in YPD, with the final culture grown in 10 mL YPGE and harvested at mid-log phase. About 150 mg of cells (wet weight) were then washed twice with ultrapure metal-free water containing 100 μM EDTA (TraceSELECT; Sigma) followed by 2 more washes with ultrapure water to remove any residual EDTA. After washing, samples were weighed, resuspended in 65 µL of ultrapure water, and were digested by adding 175 µL of 67–70% nitric acid (TraceSELECT; Sigma) followed by heating at 90°C for 18 h. The samples were cooled and were digested again by the addition of 200 µL of 30% H2O2 (Sigma-Supelco) followed by heating at 90°C for 6 h. After cooling, the samples were then diluted to 8 mL in ultrapure water and analyzed. Copper standard solutions were prepared by diluting commercially available mixed metal standards (BDH Aristar Plus).
Mitochondrial isolation
Crude mitochondria were isolated as described previously (Meisinger et al. 2006). Yeast cells were grown in 100–500 mL YPGE or YPGalactose media and were harvested at mid-log phase. The cell pellet (2–10 g) was incubated in DTT buffer (100 mM Tris-HCl, pH 9.4, 10 mM DTT, at 2 mL/g of cells) for 20 min at 30°C. Cells were then pelleted, washed in zymolyase buffer (1.2 M sorbitol, 20 mM potassium phosphate, pH 7.4 at 7 mL/g of cells), and were resuspended in zymolyase buffer containing 3 mg zymolyase (US Biological Life Sciences) per gram of cell pellet. The cell suspension was incubated at 30°C for 45 min. The efficiency of digestion was checked spectrophotometrically by diluting zymolyase-treated cells in water and measuring optical density at 600 nm. Spheroplasts, thus obtained, were pelleted at 3,000 × g for 5 min and were homogenized in homogenization buffer [0.6 M sorbitol, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 1 mM PMSF, 0.2% (w/v) BSA (essentially fatty acid-free, Sigma-Aldrich) at 6.5 mL/g of cells] with 15 strokes using a glass Teflon homogenizer with pestle B. The homogenate was then diluted in equal volume of homogenization buffer and was centrifuged at 1,500 × g for 5 min at 4°C. The supernatant was centrifuged again at 4,000 × g for 5 min at 4°C and the final supernatant was centrifuged at 12,000 × g at 4°C for 15 min to pellet mitochondria. Mitochondria were resuspended in SEM buffer (250 mM sucrose, 1 mM EDTA, 10 mM MOPS-KOH, pH 7.2, containing 1× EDTA-free protease-inhibitor cocktail from Roche).
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis, blue native-polyacrylamide gel electrophoresis, and western blotting
For sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)/western blotting experiments, yeast cells were grown in 10 mL YPGE or YPGalactose media and 2 × 108 cells were harvested and were lysed using alkaline lysis protocol (Buchanan et al. 2016). Briefly, cells were incubated in 400 µL of 0.2 M NaOH for 5 min and were pelleted at 18,000 × g for 30 s. The supernatant was removed completely, and the pellet was resuspended in 65 µL of 4× LDS buffer + 10 µL of 10× reducing agent. The samples were then heated at 70°C for 15 min and were pelleted at 18,000 × g for 30 s. Twenty microliters of the supernatant was used for SDS-PAGE/western blotting. Samples for mitochondrial SDS-PAGE/western blotting experiments were prepared using radioimmunoprecipitation assay (RIPA) buffer extraction. Mitochondria were pelleted and resuspended in protease-inhibitor containing RIPA buffer (at 4 µL/mg of mitochondrial wet weight) by vortexing. The mixture was incubated on ice for 30 min with intermittent vortexing followed by centrifugation at 14,000 × g for 10 min at 4°C and the mitochondrial extracts were obtained by collecting the supernatant. Protein estimation was done using Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). The samples were first denatured by the addition of LDS buffer and reducing agent followed by heating at 70°C for 10 min. Indicated amounts of the denatured extracts were then separated using NuPAGE 12% Bis-Tris gels (Thermo Fisher Scientific) and blotted onto a polyvinylidene difluoride (PVDF) membrane.
Samples for blue native-polyacrylamide gel electrophoresis (BN-PAGE)/western blotting experiments were prepared by solubilizing crude mitochondria in 1% digitonin, followed by incubation on ice for 30 min and centrifugation at 20,000 × g for 30 min. The supernatant was collected, and G-250 sample additive was added to it. The solubilized mitochondrial proteins (20 µg) were separated using 3–12% Bis-Tris NativePAGE gel (Thermo Fisher Scientific) and were transferred onto a PVDF membrane for western blotting.
All PVDF membranes were blocked for 1 h in 5% (w/v) nonfat milk dissolved in Tris-buffered saline with 0.1% (w/v) Tween 20 (TBST-milk), followed by overnight incubation with a primary antibody in TBST-milk at 4°C. Primary antibodies were used at the following dilutions: Cox1, 1:5,000 (Abcam 110270); Cox2, 1:250,000 (Abcam 110271); Por1, 1:100,000 (Abcam 110326); Pgk1, 1:50,000 (Life Technologies 459250); Anti-V5, 1:2,500 (Invitrogen R960-25); Anti-HA, 1:2,500 (Sigma H9658); Sco1, 1:500; Sco2, 1:600 and Cox17, 1:250 (from Dr. Alexander Tzagoloff); Coa6 1:1,000 (Ghosh et al. 2016). Secondary antibodies (GE healthcare) were used at 1:5,000 dilution in TBST-milk for 1 h at room temperature. Membranes were developed using Clarity Western ECL substrate (Bio-Rad, #1705060), or Clarity Max Western ECL substrate (Bio-Rad, #1705062).
Oxygen consumption rate measurements
The oxygen consumption rate (OCR) was measured using the high-resolution O2K FluoRespirometer (Oroboros) at 30°C. Yeast cells were grown in YPGE, washed, counted, and 2 × 107 cells were injected into the reaction chamber containing 2 ml of YPGE media. After the measurement of basal cellular OCR, maximal cellular OCR was measured by injecting carbonyl cyanide m-chlorophenyl hydrazine (CCCP) at a final concentration of 5 μM. Non-mitochondrial OCR was then measured following the addition of antimycin A at a final concentration of 2 μM. The non-mitochondrial OCR was subtracted from the cellular basal and maximal OCR to obtain the mitochondrial OCR values.
RNA isolation and quantitative reverse transcription PCR analysis
Yeast cells were grown in 5 mL YPGalactose media and RNA was extracted from ∼4.5 × 107 cells by glass bead beating using the RNeasy mini kit (QIAGEN). Five hundred nanograms of total RNA was used as starting material for cDNA synthesis using SuperScript IV VILO Master Mix with ezDNase Enzyme (Thermo Fisher Scientific; Cat: 11766050). Quantitative real-time PCR was performed on CFX96TM Real-Time PCR (Bio-Rad) in a 96-well plate. Twenty microliters of PCR reactions were prepared with 2× mastermix and 20× Taqman assay (COA4: Sc 04147827_s1; COX11: Sc04171386_s1; ACT1: Sc04120488_s1, Thermo Fisher Scientific). The mRNA levels were normalized to ACT1 expression levels.
Bioinformatics
The yeast (Q05809) and human (Q9NYJ1) Coa4 sequences were retrieved from UniProt. The pairwise global sequence alignment was performed using the EMBOSS needle program (https://www.ebi.ac.uk/Tools/psa/emboss_needle/ last accessed in August 2021) with default parameters (BLOSUM 62 matrix, Gap open penalty = 10 and gap extension penalty = 0.5).
Statistical analysis
GraphPad Prism software was used to plot graphs and bar charts. Statistical analyses were performed using unpaired two-tailed Student’s t-test and the level of significance was indicated as P-values in the figures. Mean ± SD values are from at least 3 biological replicates, which are defined as independent experiments performed on 3 different days each starting from a different clone.
Results
A targeted genetic suppressor screen links Coa4 to the mitochondrial copper delivery pathway
Since several CX9CXnCX9–10C motif-containing proteins involved in copper delivery to CcO subunits are localized to the IMS (Fig. 1), we hypothesized that the uncharacterized IMS-localized CcO assembly factors containing this motif are also likely involved in copper delivery to CcO. To test our hypothesis, we designed a genetic suppressor screen inspired by previous studies, which showed that the loss of function of upstream acting copper pathway genes could be suppressed by the overexpression of downstream copper pathway genes (Glerum et al. 1996; Barros et al. 2004; Ghosh et al. 2016). We selected yeast knockouts of the following poorly characterized CX9CXnCX9–10C motif-containing IMS proteins—Cox12, Cox19, Cox23, Pet191, Cmc2, and Coa4—as our query genes because they exhibit a respiratory growth defect caused by defective CcO assembly. To link our query genes to the mitochondrial copper delivery pathway, we cloned 17 target genes, which included genes that were previously annotated to the mitochondrial copper delivery pathway (Fig. 1), or encoded IMS-localized CcO assembly factors and other CX9CXnCX9–10C motif-encoding genes, in the yeast expression vectors (Supplementary Table 1). Using this library of mutants (Table 1), we designed a targeted suppressor screen that involved the rescue of respiratory growth defects of our query mutants by the overexpression of the known members of the mitochondrial copper delivery pathway as well as IMS-localized CX9CXnCX9–10C proteins.
We first validated our expression vectors by demonstrating that they were able to rescue the respiratory growth phenotype of the corresponding null mutant (Supplementary Fig. 1). We then transformed these plasmids into our query mutants and performed a quantitative liquid growth assay in glycerol-ethanol-containing media (YPGE) to identify suppressors of their respiratory growth defect. As reported previously, we observed a strong respiratory growth defect in cox12Δ, cox19Δ, cox23Δ, pet191Δ, and cmc2Δ mutants and a comparatively milder phenotype in coa4Δ mutant (Fig. 2, a–f). The transformation with a wild type (WT) copy of the respective query gene showed an expected rescue for each of the mutants but none of the expression plasmids were able to suppress the respiratory growth defect of cox12Δ, cox19Δ, cox23Δ, pet191Δ, and cmc2Δ mutants (Fig. 2, a–e). However, in the case of coa4Δ mutant, we found that COX11 specifically suppressed its respiratory growth defect (Fig. 2f). Notably, genes such as SCO1, SCO2, and COA6, which are involved in Cox2 metallation, did not rescue the respiratory growth of coa4Δ (Fig. 2f). To increase rigor, we repeated the coa4Δ screen on agar plates and obtained similar results, corroborating our findings (Supplementary Fig. 2). We further validated our screening result by independently transforming COX11 in coa4Δ cells and demonstrating its rescue of respiratory growth defect in YPGE plates (Fig. 2g). In a reciprocal experiment, we found that the overexpression of COA4 did not rescue the respiratory growth phenotype of cox11Δ mutant (Fig. 2h). This result indicates that Coa4 and Cox11 have nonredundant roles in the copper delivery pathway to CcO.
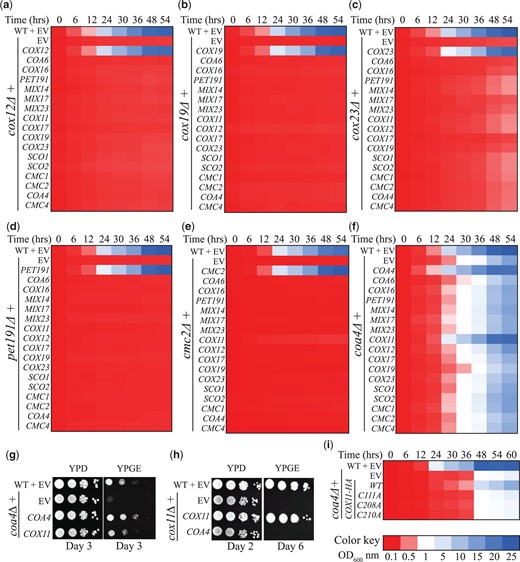
A targeted yeast genetic suppressor screen identifies Cox11 as a suppressor of the respiratory growth defect of coa4Δ mutant. a–f) Heatmap representation of growth of the query mutants cox12Δ (a), cox19Δ (b), cox23Δ (c), pet191Δ (d), cmc2Δ (e), and coa4Δ (f), transformed with the indicated plasmids in YPGE medium. Growth was initiated at a starting OD600nm of 0.1 and was monitored by measuring the absorbance at 600 nm at the indicated time points. g, h) Serial dilutions of the indicated transformants were seeded on YPD- and YPGE-containing plates and were incubated at 30°C. Images were taken on the indicated days. i) The coa4Δ mutant transformed with WT or the indicated COX11 constructs were grown in YPGE at a starting OD600nm of 0.1 and the growth was monitored by measuring the absorbance at 600 nm at the indicated time points. EV, empty vector.
Cox11 is a copper metallochaperone with 3 conserved cysteine residues that are involved in copper delivery to the CuB site on the Cox1 subunit of CcO (Carr et al. 2002; Banting and Glerum 2006; Thompson et al. 2010). Mutation in any of these cysteines completely abrogates the metallochaperone function of Cox11 and results in a respiratory growth defect (Carr et al. 2002; Banting and Glerum 2006; Thompson et al. 2010). To validate if rescue of the coa4Δ phenotype by Cox11 overexpression is through the copper metallochaperone role of Cox11, we mutated each of these conserved cysteines to alanines and found that these mutants failed to rescue the respiratory growth defect of coa4Δ (Fig. 2i). We did notice an incomplete rescue of coa4Δ mutant by Cox11-HA expression (Fig. 2i), which could be due to the presence of HA tag.
While characterizing the coa4Δ mutant, we observed that its respiratory growth defect is more pronounced at 37°C as compared to 30°C (Supplementary Fig. 3a). Surprisingly, we found that the respiratory growth at 37°C was not rescued by the episomal expression of COA4, suggesting a potential secondary mutation in this strain. A closer look at the COA4 gene locus showed that the stop codon of COA4 is located 255 bp upstream of another gene, CPR6 (Supplementary Fig. 3b), whose disruption was shown to confer heat sensitivity (Auesukaree et al. 2009). We hypothesized that construction of the null mutant of COA4 could have inadvertently disrupted the promoter region of the CPR6 gene, resulting in additional temperature sensitivity. Therefore, we constructed a new coa4Δ mutant (renamed as coa4Δd) where the gene was disrupted, instead of being completely deleted. Specifically, we replaced the sequence encoding the highly conserved twin CX9C motif of COA4 with a hygromycin cassette in its genomic locus. Unlike coa4Δ mutant, we were able to completely rescue the respiratory growth phenotype of our newly constructed coa4Δd mutant at 37°C by COA4 expressing plasmid (Supplementary Fig. 3c). Therefore, we used the coa4Δd mutant in all our future experiments.
Cox11 overexpression restores CcO biogenesis and mitochondrial respiration in the coa4Δd mutant
To understand the biochemical basis for the rescue of the respiratory growth of coa4Δ cells through Cox11 overexpression, we first measured the mitochondrial OCRs of WT and coa4Δd cells transformed with either empty vector or Cox11. Consistent with reduced respiratory growth, we found that coa4Δd has decreased basal and maximal mitochondrial OCR and Cox11 overexpression rescued these bioenergetic parameters to almost WT levels (Fig. 3a). Next, we measured the steady-state levels of the copper-containing subunits of CcO—Cox1 and Cox2—that are destabilized when copper delivery is compromised. We found a pronounced reduction in Cox1 abundance and a modest decrease in Cox2 levels in coa4Δd mitochondria (Fig. 3b, Supplementary Fig 4, a and b). Cox11 overexpression significantly rescued both Cox1 and Cox2 abundance (Fig. 3b, Supplementary Fig. 4, a and b). Analysis of steady-state levels of the respiratory chain supercomplexes showed that Cox11 overexpression also rescues the abundance of CcO (respiratory complex IV)-containing supercomplexes (Fig. 3c, Supplementary Fig. 4, c and d). These results show that Cox11 overexpression rescues activity, abundance, and assembly of CcO in coa4Δd mutant.
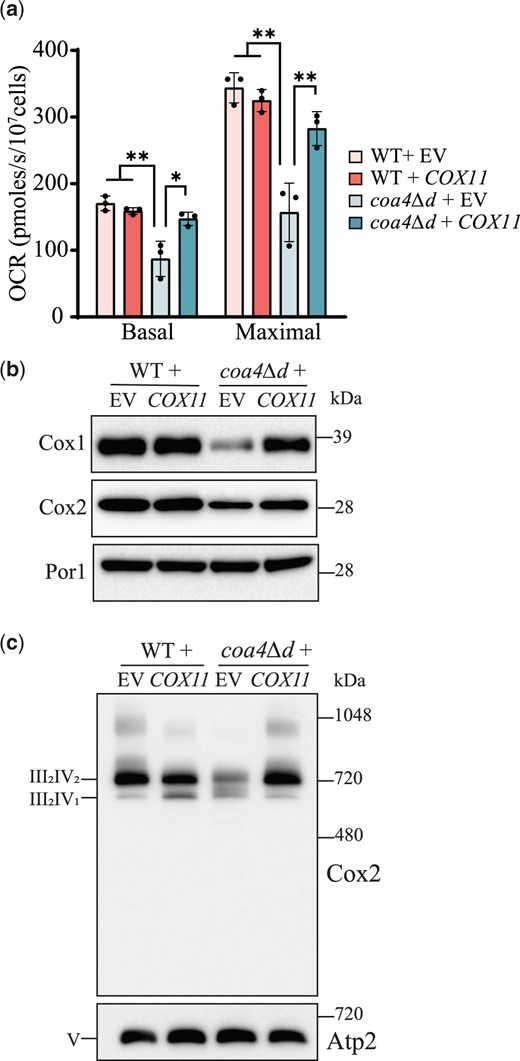
Cox11 overexpression rescues CcO function and formation in coa4Δd mutant. a) The mitochondrial OCR of the indicated transformants was measured at 30°C in YPGE. Maximal rate refers to the mitochondrial OCR after the addition of CCCP. b) SDS-PAGE/western blot analysis of Cox1 and Cox2 levels of the indicated transformants. Twenty micrograms of mitochondrial extracts were loaded in each lane. Por1, a mitochondrial outer membrane protein, was used as a loading control. c) BN-PAGE/western analysis of complex IV (CcO)-containing supercomplexes using Cox2 antibody. Twenty micrograms of digitonin solubilized mitochondria of the indicated transformants were loaded per lane. Complex V (Atp2) was used as a loading control. Data are represented as mean ± SD with each data point representing a biological replicate; *P < 0.05, **P < 0.01, n = 3. The western blots are representative images of 3 independent replicates. EV, empty vector; OCR, oxygen consumption rate.
The abundance of Coa4 and Cox11 proteins is reciprocally regulated
In order to understand how the overexpression of Cox11 is able to bypass the function of Coa4, we considered the possibility that Coa4 is required for maintaining the steady-state levels of Cox11. To investigate this, we transformed cox11Δcoa4Δ mutant with plasmids expressing Cox11-HA and/or Coa4-V5 and determined the steady-state levels of each protein in the presence or absence of the other. Surprisingly, we found that the levels of Coa4-V5 increased in the absence of Cox11 and that the levels of Cox11-HA increased in the absence of Coa4 as compared to their levels when both the proteins are expressed (Fig. 4a, Supplementary Fig. 5, a and b). This indicates that the steady-state levels of Coa4 and Cox11 are reciprocally regulated. To determine if the increase in Cox11 abundance in the absence of Coa4 is mediated at the level of mRNA abundance, we measured the expression of COX11 transcript in the absence of COA4 and found that it was not altered (Fig. 4b and Supplementary Fig. 5c). We also measured the expression of COA4 transcript in the absence of COX11 and found that it too was not altered (Supplementary Fig. 5d). Furthermore, we find that COX11 overexpression did not alter COA4 expression (Supplementary Fig. 5e), whereas in a reciprocal experiment, we see only a marginal increase in COX11 mRNA (Supplementary Fig. 5f). Notably, overexpression of COA4 and COX11 in WT cells led to a ∼4x increase in their respective mRNA levels as expected for pRS41X-series plasmids, which are maintained at 2 copies/cell (Karim et al., 2013). To determine if the reciprocal regulation between Coa4 and Cox11 protein abundance was specific, we determined the steady-state levels of other proteins that are directly involved in the copper delivery pathway to CcO—Cox17, Sco1, Sco2, and Coa6. Unlike Coa4 and Cox11, the antibodies against endogenous Cox17, Sco1, Sco2, and Coa6 were available and were thus used in this experiment. We found that the levels of these proteins did not vary in the absence or presence of either Cox11 or Coa4 (Fig. 4c, Supplementary Fig. 5, g and h). These results suggest a specific post-transcriptional regulatory cross-talk between Coa4 and Cox11.
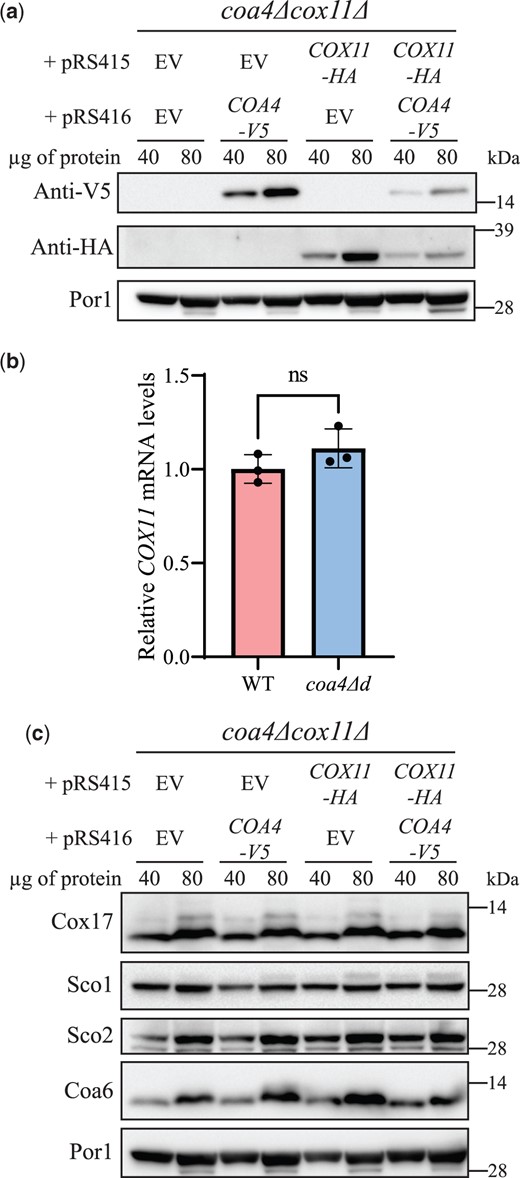
The steady-state levels of Coa4 and Cox11 are reciprocally regulated. a) SDS-PAGE/western blot analysis of the steady-state levels of V5-tagged Coa4 and HA-tagged Cox11 in cells grown in media containing 2% galactose + 0.05% glucose as carbon sources. Forty and 80 µg of mitochondrial protein extracts of the indicated transformants were loaded per lane. Por1 was used as a loading control. b) COX11 mRNA levels in WT and coa4Δd cells were measured by quantitative reverse transcription PCR. Cells were grown in YPGalactose media for 15 h before isolating RNA. The data are normalized to ACT1 expression and represented as mean ± SD (n = 3). c) SDS-PAGE/western analysis of the steady-state levels of Sco1, Sco2, Cox17, and Coa6 in the indicated transformants. The western blots are representative images of 3 independent replicates. EV, empty vector; n.s., not significant.
Copper supplementation rescues the respiratory defect of coa4Δd cells
In addition to genetic interaction studies, rescue of respiratory growth by copper supplementation has been utilized as a tool for linking genes to the mitochondrial copper delivery pathway (Ghosh et al. 2014; Garza et al. 2021). Interestingly, a previous study had reported reduced mitochondrial copper levels in coa4Δ mutant (Bestwick et al. 2010). These studies led us to test whether exogenous copper supplementation can rescue the respiratory growth defect of coa4Δ cells. Toward this end, we performed growth assays in respiratory media and found that copper supplementation can partially rescue the respiratory growth of coa4Δd cells (Fig. 5a). Notably, we found that the rescue is specific to copper supplementation because supplementation with other metals such as Fe, Mg, and Zn, which are also present in the fully assembled CcO, did not alleviate the respiratory growth defect of coa4Δd cells (Fig. 5a). Importantly, chelating bioavailable copper in the media with bathocuproinedisulfonic acid (BCS) inhibited the respiratory growth of coa4Δd cells, whereas the growth of WT cells at the same concentration of BCS was largely unaffected (Fig. 5b). Furthermore, BCS supplementation also partially abrogated the rescue of coa4Δd by COX11 overexpression (Supplementary Fig. 6a). In order to understand the physiological basis for the rescue of growth of coa4Δd cells by copper supplementation, we measured the mitochondrial OCR with and without copper supplementation and observed a partial rescue of both basal and maximal OCR of coa4Δd in copper supplemented cells (Fig. 5c). Consistent with this observation, we found that copper supplementation also partially rescued the steady-state levels of Cox1 and Cox2 subunits and the levels of the respiratory chain supercomplexes (Fig. 5, d and e, Supplementary Fig. 6, b and c). Next, in order to understand the copper deficiency phenotypes of coa4Δd cells, we measured the whole-cell copper content of WT and coa4Δd with and without copper supplementation and found that the cellular copper levels of coa4Δd were 74.4% of that of WT (Fig. 5f). Exogenous copper supplementation increased the copper levels of WT and coa4Δd to 182.8% and 171.2%, respectively (Fig. 5f). Taken together, these results suggest that Coa4 plays a role in cellular and mitochondrial copper homeostasis.
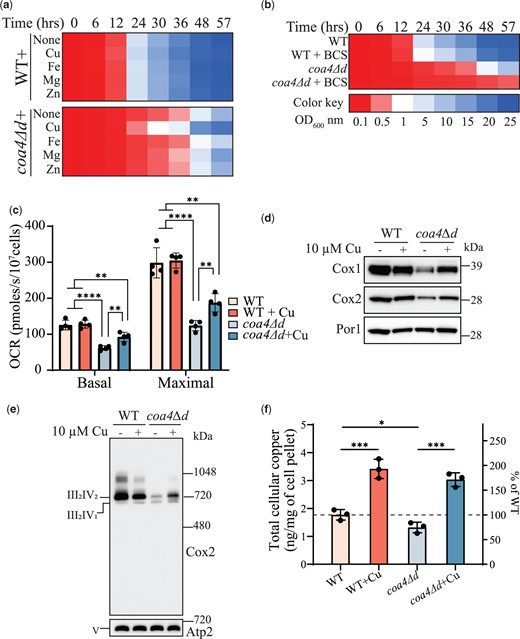
Copper supplementation rescues the respiratory defect of coa4Δ mutant. a, b) WT and coa4Δd cells were inoculated in YPGE at a starting OD600nm of 0.1 and were grown at 30°C. The growth was monitored by measuring the absorbance at 600 nm. Cu, Fe, Mg, Zn, and copper chelator, BCS were added at a final concentration of 10 μM. c) WT and coa4Δd cells were grown in YPGE at 30°C with or without copper supplementation and mitochondrial OCR was determined at 30°C in YPGE media. Maximal rate refers to the mitochondrial OCR after the addition of CCCP. d) SDS-PAGE/western blot analysis of Cox1 and Cox2 subunits of WT and coa4Δd cells grown in YPGE at 30°C with or without copper supplementation. Twenty micrograms of mitochondrial protein extracts were loaded per lane. Por1 was used as a loading control. e) BN-PAGE/western blot analysis of complex IV (CcO)-containing supercomplexes. WT and coa4Δd cells were grown in YPGE at 30°C with or without copper supplementation. Twenty micrograms of digitonin solubilized mitochondria were loaded per lane. Complex V (Atp2) was used as a loading control. f) Whole-cell copper levels of WT and coa4Δd cells were measured in the absence and presence of copper supplementation by ICP-MS. Data are shown as mean ± SD with each data point representing a biological replicate; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n ≥ 3. The growth assays and western blots are representatives of 3 biological replicates.
The function of Coa4 is evolutionarily conserved
Yeast and human Coa4 share a sequence identity and similarity of 22.3% and 44.7%, respectively. Importantly, the twin CX9C motif is conserved between the 2 proteins (Supplementary Fig. 7a). Therefore, we asked if the human COA4 can replace the function of yeast Coa4. To this end, we cloned codon-optimized human COA4 downstream of the putative yeast COA4 promoter (Fig. 6a). SDS-PAGE/western blot analysis of coa4Δd cells transformed with V5-tagged human COA4 (hsCOA4-V5) showed that it is expressed at an apparent molecular weight of ∼12.5 kDa (Fig. 6b). Growth analysis found that both hsCOA4 and hsCOA4-V5 can rescue the respiratory growth defect of coa4Δd cells (Fig. 6c). Consistent with this observation, we found that hsCOA4-V5 localizes to the mitochondria and both hsCOA4 and hsCOA4-V5 can rescue the steady-state levels of Cox1 and Cox2 subunits of CcO, indicating that the function of Coa4 is evolutionarily conserved (Fig. 6d, Supplementary Fig. 7, b and c).
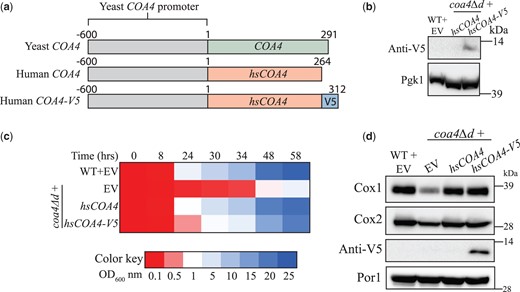
The function of Coa4 is evolutionarily conserved. a) A schematic representation of yeast COA4, human COA4, and human COA4-V5 along with the promoter used for their expression. b) SDS-PAGE/western blot analysis of the V5-tagged hsCOA4 levels in the indicated transformants. Cells were grown in YPGE at 30°C. Twenty microliters of the protein extracts from 2 × 108 cells were loaded per lane. Pgk1, a cytosolic protein, was used as a loading control. c) The indicated transformants were inoculated in YPGE at a starting OD600nm of 0.1 and were grown at 30°C. The growth was monitored by measuring the absorbance at 600 nm. d) SDS-PAGE/western blot analysis of Cox1, Cox2, and V5-tagged hsCOA4 levels in mitochondria isolated from the indicated transformants. Cells were grown in YPGE at 30°C to log phase before mitochondrial isolation and 80 µg of mitochondrial protein extracts were loaded per lane. All data are representatives of 3 biological replicates. EV, empty vector.
Discussion
Biogenesis of CcO is a complex process requiring a large number of assembly factors, including a number of twin CX9C proteins present in the mitochondrial IMS (Longen et al. 2009). Although discovered more than a decade ago, the roles of some of these twin CX9C proteins in CcO biogenesis remain obscure. The first step in understanding their functions requires linking them to the known processes and pathways of CcO biogenesis via genetic or biochemical interaction studies. Here, we took a genetic approach to link Coa4, a twin CX9C protein of the mitochondrial IMS, to the mitochondrial copper delivery pathway to CcO. Specifically, we showed that overexpression of Cox11, a metallochaperone required for copper delivery to CcO subunit Cox1, can rescue the respiratory defects of coa4Δ mutant. Additionally, we demonstrated that copper supplementation can ameliorate respiratory defects of coa4Δ mutant. Our study, thus, links Coa4 function to mitochondrial copper biology and places it in the mitochondrial copper delivery pathway to the Cox1 module of CcO.
Coa4, originally named Cmc3, was identified in a systematic screen of twin CX9C proteins and was shown to be required for mitochondrial respiration (Longen et al. 2009). A subsequent study showed that Cmc3 is a CcO assembly factor that is specifically required for the abundance, activity, and assembly of CcO and was therefore renamed as Coa4 (Cytochrome Oxidase Assembly factor 4; Bestwick et al. 2010). In the same study, it was shown that Coa4 is not required for the translation of Cox1 but acts in the later stages of Cox1 maturation (Bestwick et al. 2010). Specifically, it was shown that Coa4 acts downstream of Shy1 (Bestwick et al. 2010), a mitochondrial inner membrane protein that is required for heme a addition into Cox1 (Mashkevich et al. 1997; Smith et al. 2005; Khalimonchuk et al. 2010). Data from these previous studies are in agreement with our study, which suggests that Coa4 acts in the later steps of Cox1 assembly, specifically in the insertion of copper by Cox11.
Cox11 is an evolutionarily conserved copper metallochaperone that is essential for copper delivery to the CuB site in the Cox1 subunit of CcO (Hiser et al. 2000; Carr et al. 2002). The metallation of Cox1 with copper is preceded by the insertion of heme cofactors requiring the coordination of the heme biosynthesis machinery with the copper insertion machinery (Timon-Gomez et al. 2018). Mutations that disrupt copper insertion leads to the accumulation of a pro-oxidant Cox1 intermediate, which contains redox-active heme groups that generate deleterious reactive oxygen species (ROS; Khalimonchuk et al. 2007). Consistently, it has been shown that coa4Δ cells exhibit increased ROS production and reducing agents, such as DTT or reduced glutathione (GSH), can rescue the respiratory growth of coa4Δ mutant by quenching the ROS (Bode et al. 2013). Interestingly, the rescue of the respiratory growth of coa4Δ cells by DTT or GSH occurred without the restoration of CcO assembly (Bode et al. 2013). In contrast, Cox11 rescue of respiratory growth of coa4Δd cells accompanied the restoration of the abundance, activity, and assembly of CcO (Fig. 3). Moreover, cysteine mutants of Cox11 that are incapable of binding copper, failed to rescue the respiratory defect of coa4Δd (Fig. 2i). These results show that Cox11-based rescue of Coa4 deficiency is not simply due to suppression of ROS but rather restored copper delivery to Cox1.
Our finding that only Cox11 can rescue coa4Δ and not the other way around rules out their overlapping roles and suggests that Coa4 acts upstream of Cox11 in the copper delivery pathway. Our initial attempts to detect protein:protein interaction between Cox11 and Coa4 via coimmunoprecipitation/mass spectrometry experiments were not successful (data not shown). In the absence of a direct evidence for physical interaction between these proteins, the most likely explanation for this genetic interaction is that they are part of a common pathway. Our finding raises an obvious question—what is the biochemical role of Coa4 in the copper delivery pathway? Since deletion of Coa4 did not lead to a complete abrogation of CcO biogenesis and activity, it is unlikely that Coa4 functions as a copper metallochaperone. This is because prior studies with bonafide copper metallochaperones—Cox17, Sco1, and Cox11—of the mitochondrial copper delivery pathway have shown that their deletion leads to a complete abrogation of copper delivery to CcO, which results in much more severe phenotypes (Heaton et al. 2000; Carr et al. 2002; 2005; Horng et al. 2005). Importantly, Coa4 lacks the copper-binding cysteine motif that is found in Cox17, further negating its metallochaperone role. We speculate that Coa4 facilitates Cox1 metallation through the regulation of Cox11 by directly or indirectly impacting the delivery and/or binding of copper to Cox11.
A previous study had reported that coa4Δ cells have decreased levels of mitochondrial copper (Bestwick et al. 2010), which could be explained by our finding that coa4Δd mutant has reduced cellular copper levels (Fig. 5f). This implies that Coa4 plays a role in the maintenance of cellular copper levels. A similar cellular copper deficiency phenotype has been reported in human patient fibroblasts with mutations in genes encoding CcO assembly factors such as SCO1, SCO2, COX10, and COX15 (Leary et al. 2007) and in the tissue-specific SCO1 knockout mice (Hlynialuk et al. 2015; Baker et al. 2017). The mechanism underlying reduced cellular copper content in these mammalian cells has been attributed to either increased copper efflux or decreased copper import due to an increased turnover or mislocalization of CTR1, a plasma membrane-localized copper transporter (Leary et al. 2007; Hlynialuk et al. 2015; Baker et al. 2017). Since we found that the function of Coa4 is evolutionarily conserved (Fig. 6), it would be interesting to see if our observation holds true in higher model organisms.
In summary, we conclude that the respiratory deficiency phenotype of coa4Δd is a direct result of 2 different aspects of copper biology and that Coa4 could have 2 distinct but closely related roles in the assembly of CcO: (1) Coa4 affects the assembly of CcO through its role in copper metallation of Cox1 and this function of Coa4 could be bypassed by the overexpression of the Cox1-specific copper metallochaperone Cox11; (2) Coa4 is required for the maintenance of intracellular copper levels and indirectly affects the assembly of CcO by influencing copper availability to the mitochondrial copper delivery pathway.
Data availability
Yeast mutants and all plasmids reported in this manuscript are available upon request. The authors affirm that all data necessary for confirming the conclusions of the article are present within the article, figures, tables, and supplementary material.
Supplemental material is available at GENETICS online.
Acknowledgments
We thank Prof. Alexander Tzagoloff for generously providing Cox17, Sco1, and Sco2 antibodies, Dr. Natalie Garza for assistance with ICP-MS experiments, and Dr. Sagnika Ghosh for proofreading this manuscript. We also thank Profs. Miriam L. Greenberg, Oleh Khalimonchuk, and Antoni Barrientos for providing yeast strains.
Funding
This work was supported by the National Institutes of Health award R01GM111672 to VMG and the Welch Foundation Grant (A-1810) to VMG. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of interest
The authors declare that there is no conflict of interest.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}